Adrenal Gland Disorders: Cushing´s Syndrome, Conn’s Syndrome, Addison’s Disease, and Phaeochromocytoma
Table of Contents
Synopsis: The Adrenal Glands
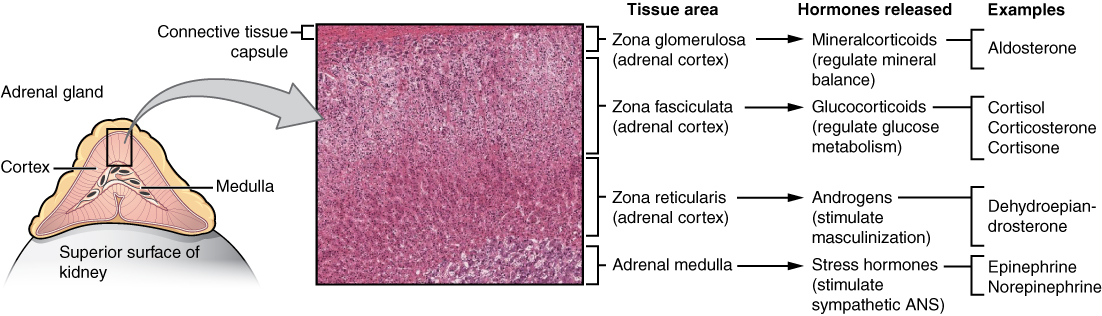
The adrenal glands are a central organ or various hormonal metabolic cycles. The adrenal medulla corresponds to a peripheral sympathetic ganglion and is responsible for the production of adrenaline and noradrenaline. The adrenal cortex consists of three anatomically and functionally different layers and is responsible for producing mineralocorticoids (aldosterone), glucocorticoids (cortisol) and androgens (dehydroepiandrosterone, androstenedione).
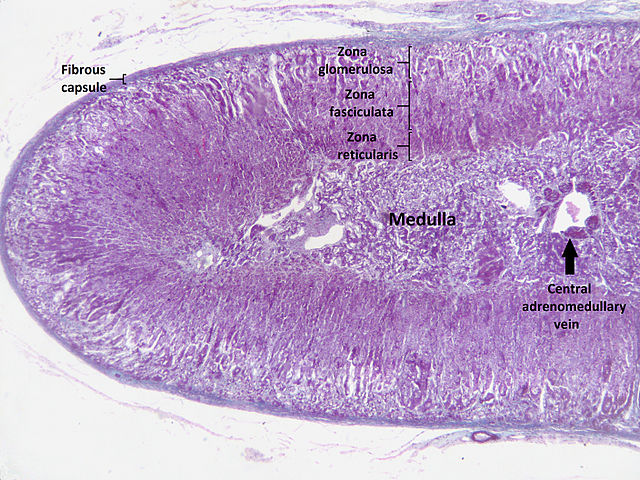
Image: „Digital camera shot through a microscope; human adrenal gland.” by Jpogi. License: Public Domain
The synthesis and secretion of these various hormones are performed by a complex system of different enzyme cascades and functional circuits. The renin-angiotensin-aldosterone system (RAAS) and the neuronal control centers (hypothalamus and pituitary gland) are two of these circuits. It is easy to imagine that disorders in these finely tuned systems could induce a variety of illnesses.
Hypercortisolism (Cushing´s Syndrome)
Cortisol compounds are widely used medications in modern medicine. Many people are prescribed long-term, regular administration. It is thus easy to infer that hypercortisolism is a relatively common disease.
Etiology of hypercortisolism
In the etiology, we differentiate between exogenous hypercortisolism stemming from long-term treatment with glucocorticosteroids, and an endogenous increased production and secretion of cortisol or ACTH. Yet in all cases, the result is hypercortisolism with subsequent Cushing’s syndrome. Exogenous hypercortisolism is the most frequent cause. With the endogenous Cushing’s syndrome, one must differentiate between the ACTH-dependent variety and the ACTH-independent variety.
If an ACTH-producing adenoma exists in the anterior lobe of the pituitary gland or a primary hypothalamic hyperfunction, central Cushing’s syndrome is present, also known as M. Cushing. This is the largest group of the endogenous manifestations of Cushing’s syndrome.
- The ACTH may also be increased ectopically, paraneoplastically via small-celled pulmonary carcinoma, or carcinoids
- A (reversible) increase in ACTH is also facilitated by alcohol consumption
- ACTH-independent Cushing’s syndrome can be caused by a cortisol-producing adenoma or carcinoma of the adrenal cortex.
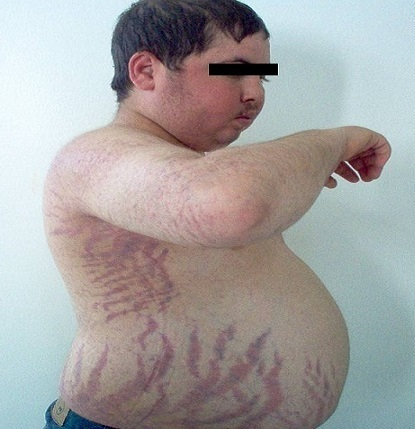
Symptoms of hypercortisolism
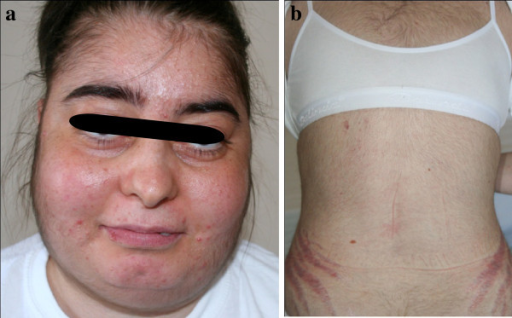
The clinical presentation of the patient stems from the manifold effects of cortisol. The impact on the lipometabolism is very impairing. This results in central obesity with a bull neck and moon face due to fat redistribution. The skin exhibits poor wound healing with a tendency for acne, stretch marks, atrophy, ulcers and boils.
There is a catabolic protein metabolism with osteoporosis (and increased risk of fracture or osteonecrosis), myopathy and adynamia. This also provokes a diabetic metabolic state. This may also cause or worsen arterial hypertension. If an ACTH-dependent form is present, the androgens may also be increased: Women experience virilism with hirsutism and menstrual disruptions.

Cushingoid symptoms
Diagnosing hypercortisolism
The symptoms or medication history lead to a suspected diagnosis. A low-dosage dexamethasone inhibitor test is conducted as an initial test of effective hypercortisolism: 2 mg dexamethasone are administered in the late evening. This should actually result in suppression of endogenous cortisol production the next morning. If this does not work, hypercortisolism is present, which must then be further investigated for its etiology:
- The corticotropin-releasing hormone (CRH) test: ACTH concentration is measured before and after administration of CRH. Hypothalamic hyperfunctions and pituitary adenomas (= M. Cushing) undergo an increase in ACTH after CRH administration.
- The high-dosage dexamethasone inhibitor test: 8 mg dexamethasone is administered at midnight for two days. This can achieve a suppression of cortisol with central Cushing’s syndrome. If an adrenal tumor or ectopic cortisol production is present, this does not work.
Imaging of the sella or adrenal glands via CT or MRT are also helpful. Paraneoplastic ACTH increases sometimes entail an increase in the metabolite lipotropin.
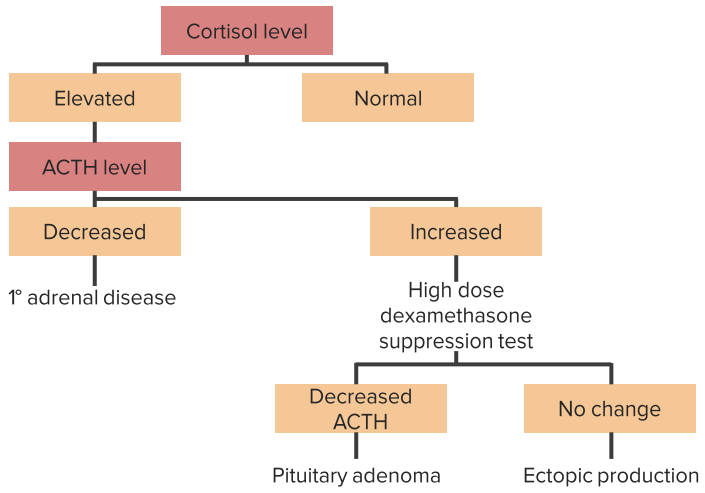
Stepwise approach to diagnose Cushing´s syndrome
Differential diagnosis
Along with the many causes of “true” hypercortisolism, one must consider that cortisol can also be increased as a stress hormone with various psychiatric symptoms (e.g. depression). Not every adrenal tumor that happens to be discovered necessarily entails symptoms of Cushing’s syndrome. Varieties that do not produce hormones are called incidentalomas.
Treating hypercortisolism
The treatment is oriented toward the cause of the hypercortisolism. Hormonally active adrenal tumors can be easily removed with a minimally invasive laparoscopic adrenalectomy. The other side takes on the role of hormone production, although glucocorticoid substitution may be necessary for a period of time. Central Cushing’s syndromes can also be treated surgically. Radiation therapy is also useful.
Inoperable adrenal cortex carcinomas or paraneoplastic ACTH syndromes can be treated symptomatically by a blockage of cortisol production. Various compounds are available for this (ketoconazole, octreotide, metyrapone, aminoglutethimide).
Conn’s Syndrome (Hyperaldosteronism)
Conn’s syndrome describes a primary hyperaldosteronism. It causes increased production of aldosterone within the adrenal cortex.
Etiology of Conn’s syndrome
In most cases, there is an idiopathic hyperplasia of the zona glomerulosa, and this may be uni- or bilateral. More uncommonly, an aldosterone-producing adenoma may be present. This often entails a much greater number of symptoms. Causal carcinomas are rare.
Primary and secondary hyperaldosteronism
Symptoms of Conn’s syndrome
Symptoms are usually relatively mild, with arterial hypertension and a normokalaemic metabolic state. Should secondary hypertension be suspected, Conn’s syndrome should always be considered as this is responsible for about 5—10 % of cases.
The distinct, but more uncommon symptoms (often coupled with the adenomas) consist of hypertension with headaches and potential organ damage, hypokalaemia with muscle weakness, constipation, EKG changes and polyuria, as well as alkalosis.
Diagnosing Conn’s syndrome
The symptoms must be indicative of Conn’s syndrome. Lab chemical tests accordingly reveal increased plasma aldosterone levels with increased aldosterone/renin quotients, and possibly hypokalaemia and/or alkalosis.
Differential diagnosis
Other causes that determine hyperaldosteronism include sodium deficiency or renal ischemia. Limited liver function or mutated renal transport channels (Bartter syndrome or Gitelman syndrome) also lead to secondary increased aldosterone levels.
Treating Conn’s syndrome
Adenomas can now be removed with a minimally invasive laparoscopic adrenalectomy. The idiopathic hyperaldosteronism is symptomatically treated with spironolactone and perhaps potassium-sparing diuretics. Carcinomas should be operatively and chemotherapeutically supplied after staging. Its prognosis is bad.
Adrenal Cortex Insufficiency
Insufficiency of the adrenal cortex results in the failure of hormonal regulatory circuits with the correspondingly marked clinical presentation. The causes and forms thereof are manifold.
Etiology of adrenal cortex insufficiency
Adrenal cortex insufficiency primarily forms from a disruption in the function of the adrenal cortex. This may be localized or be part of a generalized disease. Addison’s disease entails an autoimmune reaction with the destruction of the adrenal cortex tissue, during which the autoantibodies often target the 17 alpha-hydroxylase. One famous patient of this disease was John F. Kennedy.
Furthermore, carcinomic metastases are possible, often with pulmonary carcinomas, melanomas or kidney cell carcinomas. Infectious diseases can also damage the adrenal cortex. For instance, tuberculosis plays a significant role. Along with these rather chronic forms, acute failures of the adrenal cortex can also occur.
Important for the symptoms, but also especially frequent in exams, is Waterhouse-Friedrichsen syndrome as an expression of hemorrhagic infarction of the adrenal cortex as part of a meningococcal infection. Other reasons for acute bleeding may also damage the adrenal cortex, such as Marcumar therapy, operative complications, trauma or septicemia.
The organism attempts to counteract a primary adrenal cortex insufficiency with increased ACTH levels. ACTH consists of the precursor of proopiomelanocortin, which branches off into the melanocyte-stimulating hormone (MSH) and endorphins.
Secondary damage chiefly stems from an ACTH deficiency caused by insufficiency of the neuronal regulatory organs (pituitary gland or hypothalamus) or long-term treatment with corticosteroids.

Symptoms of adrenal cortex insufficiency
The range of symptoms is diversified and depends on the level of destruction in the adrenal cortex. Every stage from latent adrenal cortex insufficiency and a lack of symptoms up to endocrine coma are possible. People with a latent (and unknown) adrenal cortex insufficiency are the most vulnerable.
Specific strain factors may lead to acute decompensation of the hormonal situation: Addisonian crisis. Along with the aforementioned symptoms, highly different symptoms may also arise exsiccosis, hypotonic shock, pseudo-peritonitis, diarrhea and vomiting, hypoglycemia with metabolic acidosis, and eventual delirium or even endocrine coma.
The compensatory increase of MSH levels can also result in darker skin pigmentation in individuals with primary adrenal cortex insufficiency.
Diagnosing adrenal cortex insufficiency
First, it is necessary to determine the ACTH level in the plasma. This increase is typical of Addison’s disease due to the compensatory efforts of the regulatory circuits. By definition, secondary adrenal cortex insufficiencies entail lower ACTH-plasma levels. Also, no increase in the ACTH level is possible with the CRH test.
The ACTH test is also used. With this, the course of the serum cortisol level is observed before and after administrating ACTH. Based on the pathophysiological considerations, an increase in cortisol after stimulation by ACTH with a primary adrenal cortex insufficiency is not possible. If secondary adrenal cortex insufficiency is present, stimulation with ACTH is still possible and increased cortisol levels can be observed.
Imaging procedures may be used to depict potential structural damage to the adrenal cortices as well as the search for autoantibodies in the case of Addison’s disease, for purposes of further aetiological clarification. It is also wise to monitor electrolyte levels.
Differential diagnosis
Admittedly, the symptoms of adrenal cortex insufficiency may be largely non-specific. One must consider other reasons for weight loss or weakness. Abdominal illnesses associated with diarrhea, vomiting or peritonitis must be taken into consideration. Potential effects of medication are especially possible with electrolyte disorders. If an Addisonian crisis develops, all of the differential diagnostics for shock or acute abdomen are helpful and must be conducted systematically.
Treating adrenal cortex insufficiency
The hormone deficiency must be compensated. The hormonal circuits are highly complex. The physiological hormone states must frequently adjust to various situations, and they vary greatly. Infections, operations, and other bodily strains especially entail increased cortisol levels. Extensive training is an absolutely necessary part of the treatment concept in order for the patients to receive adequate hormone substitution.
The following substitutions are useful:
Glucocorticoids should be administered throughout the day according to the physiological secretion, whereby the highest daily dosage is taken in the early morning. The daily dosage amounts to about 15—25 mg hydrocortisone, can be selected individually and should be evaluated on a regular basis.
9a fludrocortisone has proven useful against mineralocorticoid deficiency. The patient’s daily requirements also vary according to the age of the patient, and medication is spread over 2—3 daily doses. The proper dosage can be determined by lab chemical electrolyte monitoring and normalized blood pressure (also under orthostatic conditions).
Every stress situation requires immediate adjustment of the dosage of hydrocortisone, i.e. the daily dosage must be increased 3 to 5 times. An effective glucocorticoid must be administered parenterally to counteract complications (e.g. vomiting).
If women experience a loss of libido, administration of dehydroepiandrosterone can be attempted.
The Addisonian crisis is a medical emergency and requires urgent treatment. Corresponding blood samples must be taken quickly to determine the current health status. This is immediately followed by calculated administration of NaCl, glucose, and hydrocortisone. Continuous monitoring of the patient is also recommended.
NaCl and glucose amend corresponding deficiencies and simultaneously counteract hypovolaemia. Amending the sodium deficiency must take place slowly to keep the risk of central pontine myelinolytic minimal.
Hydrocortisone is administered first as a bolus (100 mg IV) and then as a prolonged infusion (200 mg/24h).
In order to be able to immediately react to an Addisonian crisis, the patients must always have emergency medication available at hand. This includes, for instance, prednisolone suppositories. It is also sensible to provide them with an emergency ID.
Phaeochromocytoma
Phaeochromocytoma is a disease of the adrenal medulla. The adrenal medulla is a sympathetic ganglion made from catecholamine-producing cells. A tumor-like restructuring of these calls results in the excess production of adrenaline and noradrenaline. The patients are usually middle-aged when this disease sets in.

Etiology of phaeochromocytoma
Phaeochromocytomas are usually benign (about 85 % of cases) and unilateral (about 90 % of cases) tumors of the adrenal glands. More uncommon are extra-adrenal forms, which are more often (up to 30 %) malignant. Hereditary causes can usually be determined. This is how phaeochromocytomas form during multiple endocrine neoplasias (MEN syndrome type 2), Von Hippel-Lindau syndrome or neurofibromatosis type 1.
Symptoms of phaeochromocytoma
Phaeochromocytomas are the cause of secondary hypertension affecting about 0.2 % of all patients with hypertension. There are paroxysmal forms of hypertension due to acute hormone releases or persistent hypertonic circuit states from continuous hormone secretions.
The remaining symptoms are induced by the physiological effects of adrenaline and noradrenaline: Vasoconstrictions result in pale skin, the stress reaction leads to glucose preparation in the blood, and an overall catabolic metabolic state causes weight loss. Leukocytosis is typically found in chemical lab tests.
Diagnosing phaeochromocytoma
Phaeochromocytoma must be considered upon indications of secondary hypertension with a lack of nocturnal decrease and treatment-resistant hypertonic blood pressure values, breakouts of sweat, paleness and sudden blood pressure
crises.
The diagnosis is made upon verification of catecholamine production. Influential medications for catecholamine (e.g. sympathomimetics of asthma treatment or sympatholytics (e.g. alpha-blockers of benign prostate obstruction)) should be dismissed in advance. Catecholamine levels can be analyzed by using various methods.
Increased metabolite measurements (metanephrine and normetanephrine) in the plasma (or 24 hours of collected urine) indicate phaeochromocytoma.
The diagnosis is confirmed by the clonidine test. Physiopsychologically clonidine inhibits the synthesis of adrenaline and noradrenaline. Autonomous overproduction eludes this inhibition.
The tumors can be located via endosonographic procedures or CT/MRT imaging. The scintigraphy or single-photon emission CT (SPECT) is often used to eliminate extra-adrenal tumors.
Additional clinical, apparating or genetic testing of the aforementioned syndromes for a possible hereditary cause may be sensible with verified phaeochromocytoma.
Differential diagnosis
Phaeochromocytoma falls into the sphere of differential diagnostic considerations of secondary hypertension. Along with many other considerations, particularly hyperthyroidism must be considered. In the event of a hyperglycemic metabolic state, diabetes mellitus and hypercortisolism must be ruled out. The ingestion of various drugs may also result in similar symptoms, and amphetamine or cocaine abuse must especially be enquired.
Treating phaeochromocytoma
Removal of the tumorous structures is the treatment of choice. A minimally invasive laparoscopic adrenalectomy is often feasible. This requires a particularly cautious approach by the surgical team, as manipulations of the adrenal gland may lead to paroxysmal adrenaline release. This risk may be counteracted with a pharmacological SNS blockage before the operation. The circulatory situation and blood sugar level must be continuously monitored during and after the operation.
If operative sanitation is not possible, normalization of the catecholamine level can be attempted via pharmacological measures. Phenoxybenzamine, prazosin or alpha-methyl-p-tyrosine are helpful.
The operative results are good. However, many patients tend to relapse. Thus, regular check-up examinations are necessary.



{kind=link}
Comentários
Enviar um comentário