Pharmacodynamics: Upregulation and Downregulation, Dose-Response Curves and More
As opposed to pharmacokinetics, pharmacodynamics is what a drug does to the body. When a drug is administered, it acts on the target cell populations by means of certain receptors. There are various types of receptors, and the drug-receptor complexes can produce a specific or a variety of pharmacological effects, based on the mechanisms activated. These effects can be desirable (therapeutic) or toxic/lethal and are often dose dependent. Thus, it is crucial to learn the pharmacodynamics of the drug to arrive at not only an effective, but also a safe dose, to the individual.
Table of Contents
Are you more of a visual learner? Check out our online video lectures and start your pharmacology course now for free!
Image : “334 [meds]” by Evan Blaser. License: CC BY-SA 2.0
Important Definitions to Understand Pharmacodynamics
Receptors
A receptor is that component (macromolecule) of a cell (on or inside the cell) that interacts with the drug, and this interaction leads to a chain of events that alter the activity of the cell.
Important: Drug binds to the receptors to illicit a biological response (toxic/therapeutic).
Effectors
Effectors are molecules that act in response to the drug (or more precisely, the drug-receptor complex) and participate in the aforementioned chain of intracellular events leading to the drug’s effects.Affinity
Affinity is the measure of the strength of the bond between the drug and its receptor. Affinity of a drug to its receptor helps determine the dose of the drug: low affinity would indicate the need of a higher dose to form enough drug-receptor complexes that would lead to a significant effect (see figure).
Selectivity
Selectivity is the preference of the drug to one receptor or its subtype (as compared to other receptors or other subtypes). In other words, it is the ability of the drug to produce one pharmacological effect over others. If a drug is selective, it will preferably bind to one receptor, but it can bind to others by increasing its concentration. For example, verapamil normally blocks Ca-channels, but it can block Na-channels at high concentrations.Specificity
Specificity is the ability of the drug to bind to only one receptor. Atropine is technically specific because it acts only at acetylcholine (ACh) receptors. However, atropine is not selective because it binds to all subtypes and causes myriad pharmacological effects.Agonism
When the drug interacts with the receptor to produce a series of events leading to a pharmacological effect, it is called agonism. For example, isoprenaline is a β adrenoreceptor agonist.Antagonism
When the drug interacts with the receptor, but does not produce any series of events, thereby “blocking” the potential action (physically or chemically), it is called antagonism. For example, propranolol is a β adrenoreceptor antagonist.Antagonists can be competitive (by binding reversibly to the receptor) or non-competitive (bind irreversibly to the receptor [e.g., covalent bonds] or by allosterically changing the conformation of the protein/enzyme). The action of a competitive inhibitor can be reversed by increasing the concentration of the agonist, whereas this is not the case for non-competitive inhibitors.
Synergism
When the action of a drug is increased when given in the presence of another drug, it is called synergism. Examples are aspirin and paracetamol.Efficacy
Efficacy is the property of the drug to produce the (desired) biological effect. Efficacy is often used when comparing or evaluating drugs in clinical trials.Potency
Potency is the concentration of a drug required to produce an effect of a specified intensity.- It is usually calculated as the concentration (or dose) required to produce 50% of the drug’s maximal effect (EC>50).
- EC50 is used in in-vitro studies only. When a drug’s potency is measured in a population (animal studies or human populations), another parameter called median effective dose or ED50 is used. ED50 is the dose that produces the desired effect (typically, a quantal effect) in 50% of the population.
Excretion
Excretion is the irreversible removal of the drug from the body. Excretion mainly occurs from the liver and the kidneys, but other organs (e.g., lungs) can also be involved.Elimination
Elimination is the process of inactivation of the drug with/without actual removal from the body. Elimination can occur due to excretion or metabolism/biotransformation of the drug. Thus, while elimination is mere inactivation of the drug, excretion is the physical transfer of the (active or inactive form of the) drug from the circulation to the excretion fluids, such as urine and bile.Receptors and Effectors
Receptors
There are five basic types of transmembrane receptors, which act, after binding to the drug or the ligand, in different ways (see figures).
- Intracellular receptors: These are protein receptors that require the drug to cross the plasma membrane; therefore, the drug needs to be lipophilic. Steroids, for example, act by this mechanism.
- Transmembrane enzymes: A drug binds to the extracellular component of this receptor, which activates an enzymatic reaction in the intracellular component.
- Tyrosine kinase: When a drug binds to the extracellular component of this receptor, it leads to dimerizing of the two parts of the receptor intracellularly. This dimerization activates the tyrosine kinase enzymes, thereby leading to phosphorylation of tyrosine molecules on target proteins. Growth hormones and interferons act through JAK-STAT-kinase receptors.
- Ligand-gated ion channels: These ion channels are ligand-gated, i.e., they are closed until the receptor binds to the drug, which then allows specific ions to pass by. For example, drugs that stimulates GABA receptors on the neurons cause chloride influx (leading to hyperpolarization and thus, inhibition).
- G-protein coupled receptors: Similar to tyrosine kinase receptors, the drug-receptor binding leads to the interaction of the G-protein with the receptor. This activated G-protein then leads to the desired pharmacological response through one or a series of effector molecules or second messengers. G-protein coupled receptors are common types of receptors in the body.
Effectors
Signal TransductionA drug, when it binds to the receptor (extracellularly), acts as a “signal” for the subsequent event or events (intracellularly) that eventually leads to the pharmacological response. This transmission of signal is called signal transduction, and the intracellular chain of events that are involved in this process are called as signal transduction cascades.
A number of molecules (called second messengers) can be involved in those chain of events. The function of the cascades is to amplify the signal of the drug. As mentioned earlier, G-protein coupled receptors act via second messengers. A G-protein contains an α subunit that binds guanosine triphosphate (GTP) and the β and γ subunits that anchor the protein in the membrane.
- The α subunit dissociates with the other two after the drug-receptor binding, and the two dissociated complexes can bind to other enzymes to generate the cascades.
- A common mechanism is binding of the α subunit to the adenylyl cyclase enzyme and activating it, which causes ATP in the cell to get converted to, is cyclic AMP (cAMP). (The activation is caused by the Gs type of G-protein; Gi inhibits adenylyl cyclase.) The increased inorganic phosphate concentrations can bind to the target or other intermediate effector molecules by phosphorylation of proteins.
Upregulation and Downregulation
Upregulation
Upregulation (i.e., increase in the number) of receptors occurs when the activity of the receptor is lower than usual (e.g., due to long-term administration of an antagonist). For example, administration of beta-blockers upregulates β adrenoreceptors. Thus, if β-blockers are abruptly stopped, it can cause rebound hypertension because of the sudden stimulation of a large number of β adrenoreceptors.Downregulation
Downregulation (i.e., decrease in number) is the inverse of upregulation. It occurs due to repeated or long-term administration of an agonist. Along with downregulation, desensitization of the receptor to the drug may also occur. This is a physicochemical alteration in the receptor which makes it unresponsive to the drug; this is also called tachyphylaxis and is seen in chronic drug use, for example.
Important: The process is useful in the prevention of cell damage due to the high concentration of an agonist.
Dose-Response Curves

When the response of the drug is plotted against its dose [A], it shows a hyperbolic curve; if log of the dose (log[A]) is used, a sigmoidal curve is seen.
This curve can be used to detect the effective concentration (or dose) of the drug at which 50% of the maximal response (EC50) of the drug is obtained. As mentioned earlier, EC50 is used to calculate/compare the potency of drugs.
Emax is the (minimum) concentration at which the maximum effect of the drug is observed. At Emax, all the receptors are occupied by the drug.
Binding Curves

When the receptor binding status (% of receptors bound) is plotted against drug concentration ([A] or log[A]), it shows a similar hyperbolic (or sigmoidal) curve.
Using this graph, we can calculate the effective concentration at which 50% of the receptors are bound (Kd). Kd shows the binding affinity of the drug — a high value of Kd indicates that a high concentration of the drug is required to bind 50% of the receptors, thereby indicating low affinity.
Bmax is the maximal binding of the drug. It is the concentration, expressed as picomoles per mg protein, at which all specific receptors are bound to the drug. As a result, Bmax can be used to calculate the density of the receptor site in a particular preparation.
Spare Receptors
When an agonist binds to a receptor, it may produce a series of events that eventually lead to the pharmacological response. This series of events may amplify the drug’s effect such that only a relatively small amount of the drug-receptor complexes may be needed to lead to maximal response. In this case, when the maximal response is seen, a percentage of receptors remain unoccupied by the drug. These receptors are called spare receptors.
Spare receptors, at least theoretically, increase the sensitivity of the drug.
Toxicity, Toxicity Curves and Therapeutic Ratio
Adverse effects of a drug are usually dose-dependent. To investigate the effectiveness and toxicity of the drug, its effects are tested in a population and plotted against concentration or dose. Sigmoid curves (if log concentrations are taken on the X-axis) are obtained, and they can reflect the effects observed.For example, in the following figure, a sedative/hypnotic agent shows the sedative action at a certain concentration range and death beyond that range. The dose (or the concentration) at which the drug shows the desired effect (sedation) in 50% of the population is ED50.
The dose (or the concentration) at which the drug shows the toxic effects (e.g., respiratory depression) in 50% of the population is TD50. The dose (or the concentration) at which the drug shows the lethal effect (death) in 50% of the population is LD50. (This is used for animal experiments.)
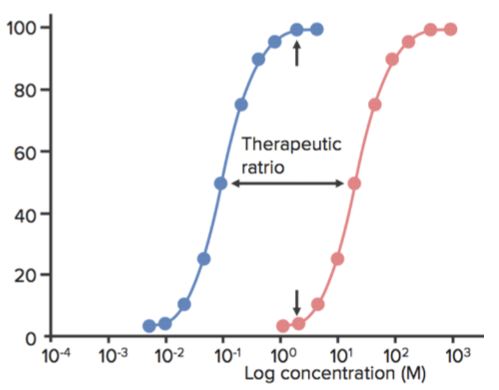
Therapeutic window is the range of dose between the minimum effective concentration and the minimum toxic concentration. Therapeutic index (TI) is defined as the ratio between the TD50 and ED50 or TI=TD50/ED50.
- This is sometimes also called as the therapeutic ratio.
- When a drug has a low TI, it means that increasing its dose can easily produce toxic (or lethal) effects.
- Tricyclic antidepressants and lithium, for example, have low TI, and their dose must therefore be monitored very carefully in patients. Imipramine, for example, can be fatal if administered in 5–6 times the maximal daily dose.
- The higher the TI, the safer the drug (example is penicillin).
Review Questions
The correct answers can be found below the references.1. Lithium has a narrow therapeutic window. The median effective serum concentration of lithium is 0.6 mEq/L and the median serum concentration at which it causes toxicity is 1.5 mEq/L. What is the therapeutic index of lithium?
- 2
- 1.5
- 2.1
- 0.9
- 2.5
- Decrease in the number of β adrenoreceptors
- Increase in G-proteins on the cell surface
- Activation of spare β adrenoreceptors
- Increase in the number of β adrenoreceptors
- Phosphorylation of most β adrenoreceptors
- Emax and EC50
- Emax and median effective dose
- Therapeutic index and quantal log dose−response curve
- Clearance and therapeutic index
- Affinity and EC50



Comentários
Enviar um comentário