Cardiomyopathy (Heart Muscle Disease) — Symptoms and Causes
Table of Contents
Image: “Heart transesophageal echocardiography (TEE) view.” by atrick J. Lynch, medical illustrator – Patrick J. Lynch, medical illustrator. License: CC BY 2.5
Definition and Classification of Cardiomyopathy
As the name says, cardiomyopathy is a myocardial disease, which comes with an impaired systolic and diastolic function. The disease affects the muscles of the heart leading to impaired function of the heart.
Two types of cardiomyopathy:
- Primary type: heart muscle disease predominantly involving the myocardium and of unknown etiology
- Genetic: hypertrophic, arrhythmogenic right ventricular dysplasia, glycogen storage disorders, mitochondrial myopathies
- Acquired: inflammatory (myocarditis), peripartum, tachycardia-induced
- Mixed: idiopathic dilated cardiomyopathy, restrictive cardiomyopathy
- Secondary type: myocardial disease of known cause/associated with a systemic disease, for example, amyloidosis, chronic alcoholic use
- Infiltrative including amyloidosis
- Storage including hemochromatosis
- Toxicity including drugs, chemotherapeutic agents
- Inflammatory including sarcoidosis
- Endocrine including rypo- or hyperthyroidism diabetes
- Neuromuscular/neurological
- Autoimmune including systemic lupus erythematosus, rheumatoid arthritis, scleroderma
The WHO classifies 5 types based on cardiac changes:
- Dilated (DCM)
- Hypertrophic non-obstructive or hypertrophic obstructive (HNCM or HOCM)
- Restrictive (RCM)
- Arrhythmogenic right ventricular (ARVC)
- So-called unclassified cardiomyopathy
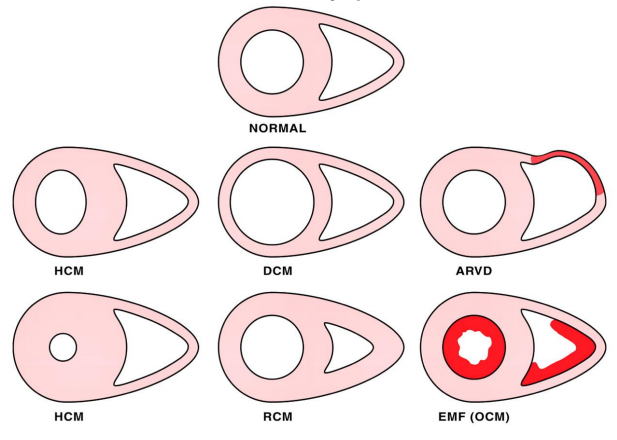
Different cardiomyopathies. Image by Lecturio.
Potential complications include heart failure, arrhythmias, and sudden death. Cardiomyopathy is the 2nd most common cause of sudden death (Ischemic heart disease is #1). In the past, few cardiomyopathies were thought to be genetic. Now, at least 1/3 are known to be genetic. Prognosis for dilated cardiomyopathy is poor, especially when diagnosed late in the course when heart failure signs and symptoms are present.
Dilated Cardiomyopathy
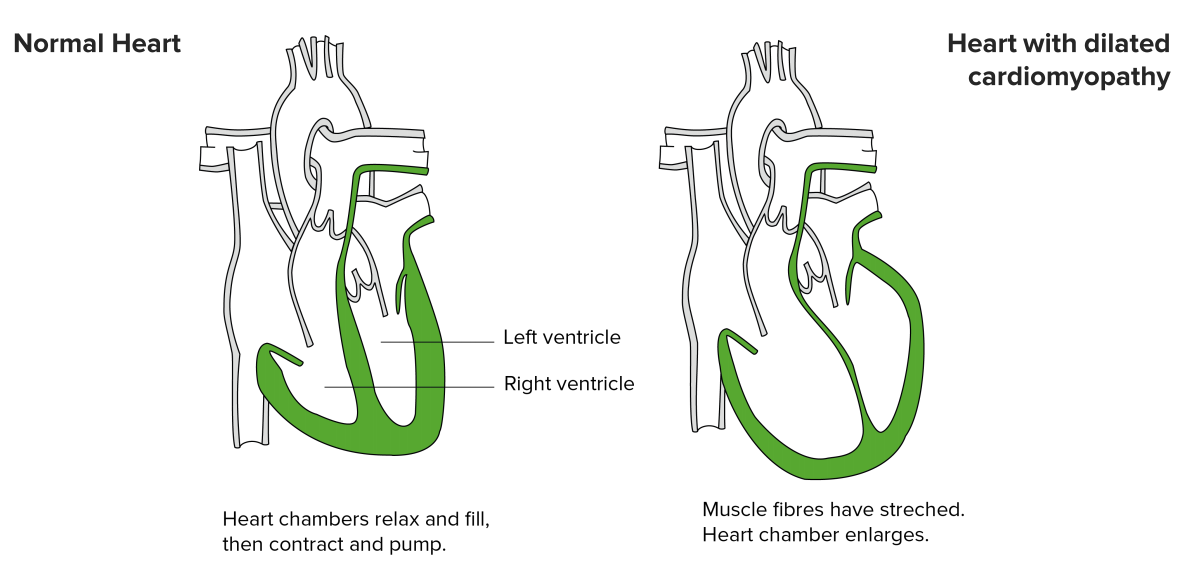
Image by Lecturio
The most common type of non-ischemic cardiomyopathy is DCM. It has an incidence of 6/100,000, which affects more men than women, is the DCM. Causal processes are usually unknown; however, it is easily identified on the basis of an enlarged, left-ventricular diameter in the echocardiography and a rarefication of cardiomyocytes in microscopic examination. It can be inherited or be a result of underlying conditions. Diseases such as diabetes, viral heart infections, thyroid disease, or abnormalities in the valves of the heart may lead to DCM. Postpartum cardiomyopathy happens in women is associated with a sudden change in cardiovascular function and blood volume after birth though the exact cause is not known.
The patients having symptoms of a heart insufficiency, like i.e. dyspnea or lung edema (left ventricular insufficiency) or blood stasis in the peripheral body (right ventricular insufficiency), should as well always raise the suspicion of a DCM.Other symptoms include pitting edema, weight gain, dizziness, fainting and shortness of breath.
ECG, blood tests, X-rays, MRI, myocardial biopsy, CT scans and cardiac catheterization are some of the tests that may help diagnose DCM cardiomyopathy.
Treatment
Consequently, it is treated (correspondent to heart insufficiency) using diuretics, vasodilators, ACE-inhibitors, and beta-blockers. This medicinal therapy helps to delay a progression of the disease and lower the risk of sudden cardiac death. It also aims at improving cardiac functions as well as removing most of the symptoms. It is necessary that a lifestyle change such as including exercises as well as changes in the diet occurs to support these medical processes as well. If arrhythmic side effects occur, the implantation of cardiac resynchronization systems is indicated additionally. If in the further course, the LVEF (left ventricular ejection fraction) falls below 35 %, also an oral anticoagulation using vitamin K antagonists such as warfarin and an ICD is necessary. However, there are asymptomatic patients as well.
Hypertrophic Cardiomyopathy
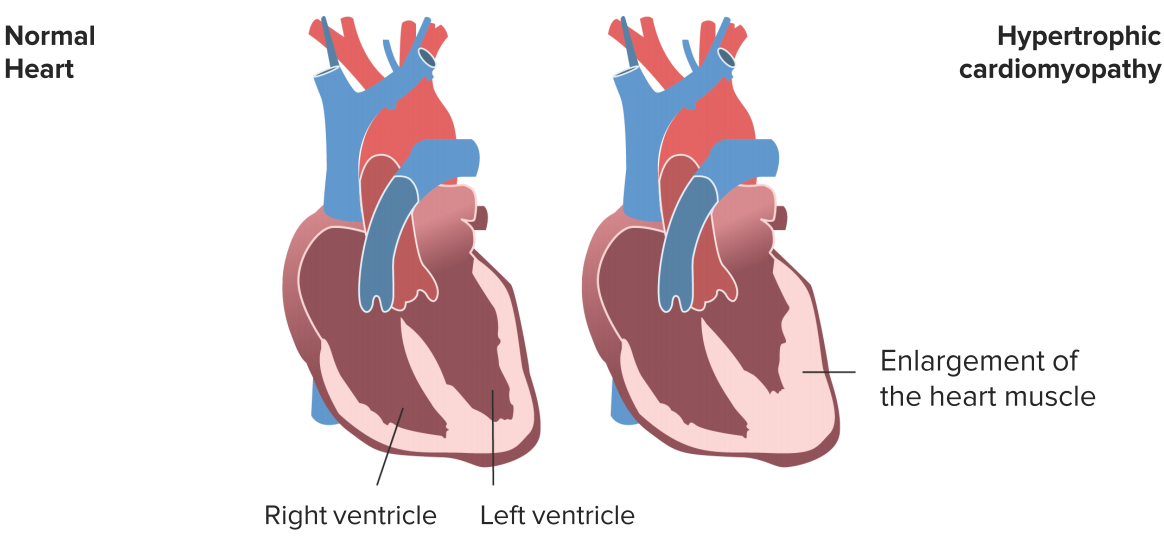
Image by Lecturio
The hypertrophic cardiomyopathy differs from the DCM in pathophysiology and etiology. In over 50 % of the cases, an inheritable mutation (autosomal dominant) can be detected, which either affects the contractile apparatus or the energy metabolism of cardiomyocytes. Subsequently, these are enlarged and disarranged. It can be diagnosed by a 2-D echocardiography, chest radiography, radionuclide imaging as well as cardiac magnetic resonance imaging.
Basic explanation is that the left ventricle of the heart has a thicker wall than usual due to mutations of the protein genes that make up the cardiac sarcomere. Symptoms may include sudden cardiac death, dyspnea, angina, palpitations, presyncope, syncope, and dizziness.
Already starting in fetuses, it is progressing at varying speed. Often, the myocardium becomes hypertrophic after puberty and the left ventricular diameter is regular or narrowed (compare DCM!), which is why the filling volume is reduced. For this reason, the HNCM is a common reason for the sudden cardiac death in young athletes.
A special feature of this type is the possible formation of a so-called dynamicmuscular bulge just below the aortic valve during systole, so that the left ventricular outflow is impaired.
Treatment of hypertrophic cardiomyopathy
- B-blockers, verapamil (calcium channel blocker), disopyramide, amiodarone have been found useful
- Beta blockers in high dose are the #1 therapy to be tried
- No pharmacological treatment is known to improve prognosis
- Atrial and ventricular arrhythmias are common and often respond to anti-arrhythmic drug therapy including beta-blockade
- Outflow tract obstruction can be improved by:
- Partial surgical resection (myectomy)
- Iatrogenic infarction of basal septum (septal ablation with alcohol)
- Implantable cardiac defibrillation (ICD) for patients with risk of sudden death
Among the named therapy approaches for DCM, at this a Morrow myectomy or an interventional TASH is expedient. The latter describes the identification of an arterial branch that feeds the bulge and a following injection of alcohol. This way muscular mass is devitalized.
Restrictive Cardiomyopathy
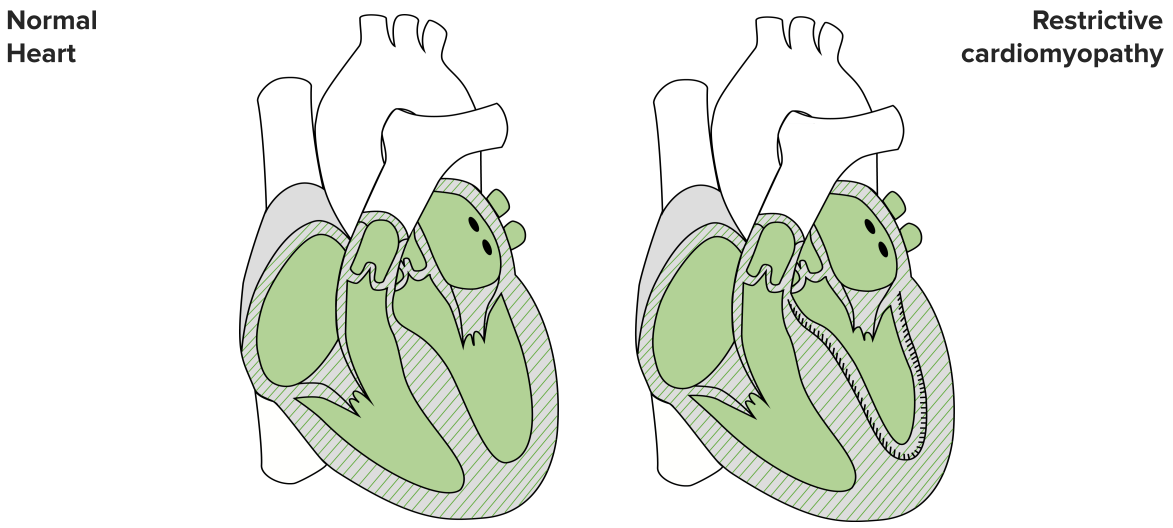
Image by Lecturio
The RCM is rather seldom in western countries, however, quite common in tropical regions. Contrary to the DCM, this is a diastolic malfunction caused by an increased rigidity (meaning reduced compliance) of the heart’s wall due to increased connective tissue, which leads to increased pressures in atria and ventricles. It is also genetical where it can be cured by using molecules that silence genes that lead to abnormal protein production.
It is hard to recognize by echocardiography, but there are also clinical symptoms of heart insufficiency. On the basis of this discrepancy, the RCM can be diagnosed.
Common causes are infiltrations of the myocardium, storage diseases, and endo-/myokarditides. Here, the Loeffler endocarditis is to be named to a special degree, which leaves scars and that way causes the rigidity. In this case, cortisone must be added to the conventional medicinal therapy. Moreover, excision of the endocardium belongs to the surgical treatments. Corticosteroids are used to treat sarcoidosis and Loeffler endocarditis. Chemotherapytreats amyloidosis. Endarterectomy treats endomyocardial fibrosis as well as Loeffler endocarditis.
Arrhythmogenic Right Ventricular Cardiomyopathy
The most uncommon type is the ARVC. This is also a genetically-induced and inheritable type of cardiomyopathies, which especially comes with ventricular arrhythmia and tachycardia. It can lead to sudden cardiac death.
Therefore, therapy includes antiarrhythmic drugs, ICD-implantation, and catheter ablation. Physical stress must be avoided. It requires changes in lifestyle as well as regular testing for people whose family member has been diagnosed with it.
It is important to understand the fundamental pathomechanisms and types of therapy. Indeed, details and numbers can leave behind a good impression; however, don’t lose yourself in details. Aside from that, the morphological differences between the different types can be memorized quite well by visualization.



{kind=link}
Comentários
Enviar um comentário