Erythema Multiforme and Stevens-Johnson Syndrome (SJS, Erythema Multiforme Major) in Children — Symptoms and Treatment
Table of Contents
- Overview
- Epidemiology of Stevens-Johnson Syndrome in Children
- Etiology and Pathophysiology of Stevens-Johnson Syndrome
- Clinical Presentation of Stevens-Johnson Syndrome in Children
- Diagnostic Workup for Stevens-Johnson Syndrome in Children
- Treatment of Stevens-Johnson Syndrome in Children
- References

Image: “Micrograph showing full-thickness epidermal necrosis with a basket weave-like stratum corneum and separation of the dermis and epidermis, skin biopsy, H&E stain.” byNephron. License: CC BY-SA 3.0
Overview
Stevens-Johnson syndrome is a form of an immune-complex-mediated hypersensitivity reaction that involves the skin and the mucous membranes with a mild to a moderate component of toxic epidermal necrolysis.
The disease can be classified into three subtypes based on the percentage of body surface area involved:
Epidemiology of Stevens-Johnson Syndrome in Children
Simple Stevens-Johnson syndrome and toxic epidermal necrolysis are considered as very rare conditions in children in western countries including the United States. The estimated incidence of Stevens-Johnson syndrome in children is approximately 1 to 7 cases per million per year while the estimated incidence of toxic epidermal necrolysis is around 0.4 to 1.5 per million per year. The condition is relatively more common in adults compared to children.
Stevens-Johnson syndrome and toxic epidermal necrolysis are more common in the whites but have no differences in the incidence between males and females.
The prognosis of Stevens-Johnson syndrome is quite good unless the skin lesions become secondarily infected. Patients with simple Stevens-Johnson syndrome have a mortality rate of around 1 to 5%, while the mortality rate of those with toxic epidermal necrolysis can be as high as 50 %. The most common cause of death in patients with Stevens-Johnson syndrome is sepsis.
Children who have malignant disease, who have tachycardia, an initial percentage of epidermal detachment of more than 10%, an elevated blood urea nitrogen above 10 mmol/L, a serum glucose level above 14 mmol/L and a bicarbonate level below 20 mmol/L have an increased risk of death if they develop Stevens-Johnson syndrome.
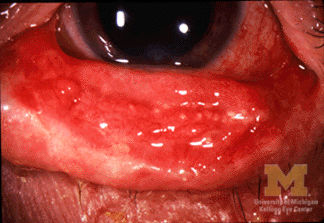
Stevens-Johnson syndrome commonly involves the eyes and survivors can develop long-term complications such as inverted eyelashes, photophobia, watery eyes, and a burning sensation in the eyes.
Etiology and Pathophysiology of Stevens-Johnson Syndrome
While Stevens-Johnson syndrome is an immune-complex-mediated hypersensitivity disorder, several etiological factors have been identified as triggers of the condition. The most common cause of Stevens-Johnson syndrome is infectious in nature. Herpes simplex virus, the human immunodeficiency virus, Coxsackie viruses, hepatitis, influenza, and mumps have been implicated with Stevens-Johnson syndrome.
Epstein-Barr virus and enteroviruses are also common etiologies of Stevens-Johnson syndrome in children. Bacterial etiologies of Stevens-Johnson syndrome include group A beta-hemolytic streptococci, brucellosis, mycobacteria, mycoplasma pneumoniae, rickettsia infections and tularemia. Typhoid fever has also been associated with an increased risk of developing Stevens-Johnson syndrome in children.
Stevens-Johnson syndrome can also be induced by non-steroidal anti-inflammatory drugs, antibiotics, phenytoin, carbamazepine, oxcarbazepine, valproate, lamotrigine, barbiturates, and allopurinol.
It is understandable that not all patients who acquire an infectious disease or take one of the previously mentioned drugs are going to develop Stevens-Johnson syndrome. This, in addition to the observation that a family history of Stevens-Johnson syndrome and toxic epidermal necrolysis is usually evident in many cases, made scientists look for possible associations between different human leukocyte antigen (HLA) subtypes and the risk of developing the syndrome.
HLA-B*1502, HLA-B*5801, and HLA-B*44 are few examples of HLA subtypes that have been linked with an increased risk of developing Stevens-Johnson syndrome.
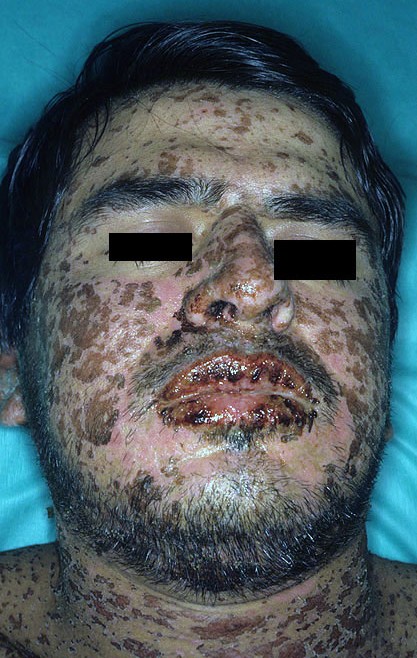
Steven Johnsons Syndrome
The exact pathogenesis of Stevens-Johnson syndrome is not fully understood. When an infectious agent or a drug metabolite is present in the immune cells, especially in patients with abnormal immune responses such as those infected with the human immunodeficiency virus, huge and complex immune particles form.
It is believed that these immune complexes are presented as antigens to tissue dendrocytes which produce tumor necrosis factor and recruit and activate T-lymphocytes.
These cytotoxic T-lymphocytes produce killer effector molecules that induce apoptosis in keratinocytes. When keratinocytes undergo apoptosis and cell death, the epidermis becomes separated from the dermis which is the hallmark of Stevens-Johnson syndrome. The dying cells increase the antigen load on the different immune cell types and trigger T-cells to produce more inflammatory cytokines and inflammation ensues.
When the skin’s integrity is impaired, the patients lose a significant amount of water, become dehydrated and can develop secondary bacterial infections of the skin lesions and sepsis. Mortality and morbidity of Stevens-Johnson syndrome are largely dependent on these pathologic changes that happen in patients who develop the condition.
Clinical Presentation of Stevens-Johnson Syndrome in Children
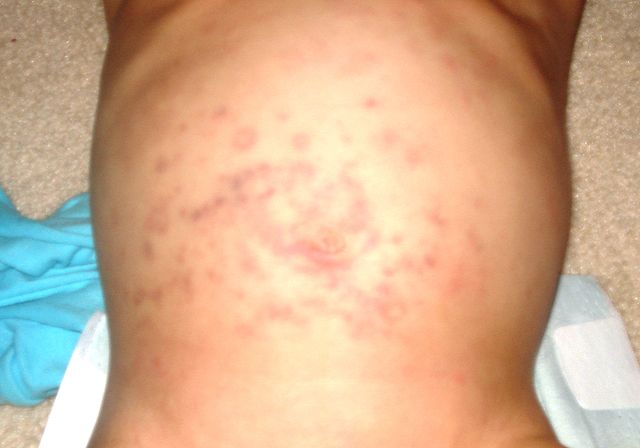
Patients usually develop a flu-like illness with malaise, muscle and joints pain, photophobia and conjunctival itching or burning sensation for one to three weeks after the introduction of one of the possible triggering drugs or the development of an upper respiratory tract infection due to one of the infectious etiologies.
After this prodromal phase, which can also be characterized by fever, patients start developing characteristic skin and mucosal lesions. Macules, large blisters, vesicles, and bullae form within the involved areas. The most affected regions are the face, thorax, scalp, palms, and very rarely the soles of the feet. These lesions are usually tender and painful to touch.
Patients with mucosal surfaces involvement can present with signs and symptoms suggestive of severe dehydration due to significant fluid loss. Edema can also develop due to protein loss from the affected skin areas. Patients can become hypothermic and can develop electrolyte imbalances which could be fatal.
More severe cases can develop acute respiratory distress syndrome and mechanical ventilation might be necessary in up to 25% of the cases. Patients can also develop diarrhea and melena due to small bowel ulcerations.
Diagnostic Workup for Stevens-Johnson Syndrome in Children
The diagnosis of Stevens-Johnson syndrome is largely based on the typical clinical findings and histologic confirmation by performing a skin biopsy. For research purposes, some scholars have pointed out that patients with Stevens-Johnson syndrome might have elevated serum levels of tumor necrosis factor alpha, interleukin-6, and C-reactive protein. Serum electrolytes should be monitored closely because these patients are known to lose excessive amounts of fluid, electrolytes and serum proteins from their skin wounds.
The emergency management of Stevens-Johnson syndrome in a child should not be delayed or dependent on the histologic findings. Regardless, a skin biopsy should be taken in any patient for the documentation of the syndrome and confirmation of the diagnosis.
The most common histologic findings seen on a biopsy taken from a bulla include minimal dermal inflammatory cell infiltrate and full-thickness necrosis of the epidermis. Sub-epidermal blisters are commonly seen. The number of keratinocytes is decreased because of apoptosis.
Immunohistochemical staining of the dermis reveals an abundance of CD4+ T-lymphocytes.The epidermis is commonly infiltrated by CD8+ T-lymphocytes.
Treatment of Stevens-Johnson Syndrome in Children
The first and most important step in the management of Stevens-Johnson syndrome in a child is to withdraw any possible offending drugs. This step has been consistently shown to improve outcome and decrease mortality.
Patients with Stevens-Johnson syndrome should be treated in a burns unit and meticulous wound care should be provided. The wounds should be debrided, moisturized, and anti-bacterial ointments should be used. Due to the excessive loss of fluid and electrolytes seen in patients with Stevens-Johnson syndrome, fluid replacement therapy should be initiated.
Patients who have severe throat and mouth blisters might find it very painful to eat. In that case, parenteral nutrition should be started but switching back to oral feeding should be made as early as possible. Patients with Stevens-Johnson syndrome can become hypothermic just like burn patients and their temperature should be managed and closely monitored.
An ophthalmologist should be consulted as early as possible to prevent long-term ocular complications which are quite common in children who survive the acute illness.
Unfortunately, corticosteroids and intravenous infusion of immunoglobulins have been used in the management of severe cases of toxic epidermal necrolysis but there are no controlled randomized clinical trials that show any benefits of these treatments.



{kind=link}
{kind=link}
{kind=link}
Comentários
Enviar um comentário