Types of Kidney Cysts (Renal Cysts) — Diagnosis and Treatment
Table of Contents
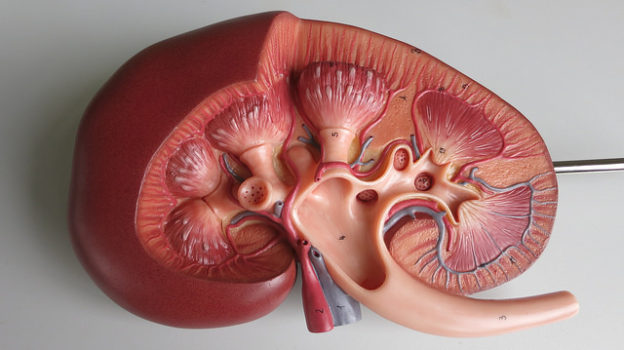
Autosomal Dominant Polycystic Kidney Disease (ADPKD)
Epidemiology of ADPKD
ADPKD is the most commonly inherited renal disease and affects approximately 1 in 800 live births. ADPKD is also responsible for 6-10% of renal therapy replacement treatments and is the leading genetic cause of end stage renal disease worldwide.
Etiology and Pathophysiology of ADPKD

Image: “Gross Pathology of Polycystic Kidneys” by Patho. License: Public Domain
Two mutations, which are inherited in an autosomal dominant fashion, are responsible for the disease:PKD1 (chromosome 16) in 85% of cases and PKD2 (chromosome 4) in 15% of cases. Nonetheless, the condition has a variable phenotypic expression which can be explained by the two-hit hypothesis, in which a second somatic mutation (second hit) appears in the normal allele and leads ultimately to cell proliferation and transverse growth, resulting in the formation of cysts that involve all parts of the nephron in the cortex and medulla.
Growth of renal cysts causes growth and deformation of the kidney, tubular obstruction and renal ischemia, leading to increased renin-angiotensin-aldosterone activity and hypertension. Kidney function progressively deteriorates, but does not decline until at least 50% of the parenchyma is destroyed.
Clinical Presentation and Symptoms of ADPKD
Abdominal or flank pain is a prominent presenting symptom in patients with ADPKD and is almost universally present. Patients also commonly complain of fatigue, breathlessness, weakness and malaise in the early stages of the disease. Hematuria is another frequent presenting manifestation, but is usually self-limited and lasts approximately one week. On physical exam, patients commonly have diastolic hypertension, which is notable for becoming less problematic as kidney function further deteriorates.
Additional findings include palpable bilateral flank masses. Symptoms that are common with kidney failure such as edema and pallor are rare on initial presentation. Extrarenal manifestations include benign cysts in the liver (94%), the seminal vesicle (40%) and the pancreas (9%), as well as connective tissue abnormalities such as colonic diverticula, mitral valve prolapse (25%), intracranial aneurysms (8%) and abdominal hernia (10%).
Laboratory Evaluation and Diagnosis of ADPKD
Ultrasound is the diagnostic modality of choice for ADPKD. It should also be used for screening family members. Other possible diagnostic imaging tests include MRA, MRI and CT-scans. The disease may also be associated with increased hematocrit due to elevated erythropoietinsecretion from the cysts.
Microalbuminuria is also common, but does not reach nephrotic-range proteinuria. Select patients with ADPKD should undergo screening for intracranial aneurysms. Indications for screening include family history of intracranial aneurysms and/or bleeding, new-onset severe headache, major elective surgery and CNS signs and symptoms.

Image: “Abdominal CT scan of an adult with autosomal dominant polycystic kidney disease. Extensive cyst formation is seen over both kidneys, with a few cysts in the liver as well.” by Steven Fruitsmaak. License: CC BY-SA 3.0
Therapy and Treatment of ADPKD
Strict blood pressure control is fundamental with either an ACE inhibitor or an ARB. Clinical studies found Vasopressin V2 receptor antagonists, such as tolvaptan, to be useful. A regular monitoring of the kidney parameters, as well as ultra sound controls, play an important part in the management of the patient.
Renal replacement therapy (dialysis) is usually required for 50% of patients above the age of 60. Nephrectomy is reserved for cases of anatomical infringement, cyst hemorrhage, infection and uncontrolled pain. A kidney transplant is the treatment of choice for patients with end stage renal disease.
Prognosis for ADPKD
50 to 75% of patients with ADPKD will require dialysis by the age of 75. Some predictors of rapid deterioration include: PKD1 mutation, large kidney size, proteinuria, male sex, and early age at diagnosis. There is no concomitant increase in renal cancer.
Autosomal Recessive Polycystic Kidney Disease (ARPKD)
Epidemiology of ARPKD
ARPKD is a childhood kidney disease with incidence of about 1/10.000 to 1/20.000 birth. It affects all racial and ethnic groups and males and females equally.
Etiology and Pathophysiology of ARPKD

Image: “Baby with distended abdomen due to voluminous kidneys” by Carsten Bergmann, Pediatric Nephrology. License: Open Access
ARPKD is caused by a mutation in the PKHD1 geneon the short arm of chromosome 6. The protein encoded by the gene is expressed by renal and hepatic epithelial cells, but its function remains only partially understood. ARPKD leads to bilateral, non-obstructive elongation of the collecting ducts, resulting ultimately in enlarged kidneys. All patients will also have congenital hepatic fibrosis, which can be more severe than the renal disease. It can lead to portal hypertension and ultimately GI hemorrhage, varices and splenomegaly.
Clinical Presentation and Symptoms of ARPKD

Image: “Potter’s phenotype with distinctive facial features” by Carsten Bergmann, Pediatric Nephrology. License: Open Access
Patients with ARPKD might present at birth with enlarged flank masses that may complicate delivery. These babies will also have Potter facies (parrot beak nose, low-set ears) and abnormal limbs. Older infants may present with abdominal distention due to renal masses or hepatosplenomegaly. Hypertension can be severe and can be the presenting symptom.
Laboratory Evaluation and Diagnosis of ARPKD
Ultrasonography is the primary diagnostic method, particularly among neonates, although a definitive diagnosis sometimes may require a biopsy. Genetic testing can also be used when clinical criteria are not met. Renin levels are usually normal.
Therapy and Treatment of ARPKD

Image: “Macroscopic appearance of advanced-stage autosomal dominant polycystic kidney disease (ADPKD) showing enlarged kidneys with multiple cysts that almost completely destroyed and replaced the renal parenchyma” by Carsten Bergmann, Pediatric Nephrology. License: Open Access
Neonatal survival is dependent on ventilation and intensive care unit treatment. Hypertension needs to be treated preferably with ACE inhibitors. Once chronic kidney disease develops, patients may require iron supplements and erythropoietin for anemia, calcium, phosphate binders and suppressors of the parathyroid to prevent metabolic bone disease, and growth hormone to fend off the effects of uremia on growth.
Unilateral or bilateral nephrectomy is performed in cases of respiratory compromise in neonates or when failure to thrive is present. Dialysis and transplantation are treatments of choice in end stage renal disease.
Prognosis for ARPKD
Prognosis varies considerably. Patients who are born with oligohydramnios most often die due to pulmonary complications. Patients who survive the neonatal period can still develop chronic kidney disease, although prognosis has improved due to renal transplants. Congenital hepatic fibrosis can still lead to significant morbidity.
Medullary Cystic Kidney Disease (MCK)
Epidemiology of MCK
MCK is a very rare genetic disorder that has been mostly reported in the United States; both genders are equally affected with no racial predilection.
Etiology and Pathophysiology of MCK
MCK presents in two types, and both are inherited in an autosomal dominant fashion. The gene responsible for type 1 is localized to chromosome 1, whereas the gene responsible for type 2 is localized on chromosome 16. Nonetheless, 16% of patients have no family history, suggesting a sporadic mutation.
The disease results in the formation of cysts in the medulla and typically presents in adulthood. These cysts may not be detected and may result in progressive kidney failure and a decrease in kidney size. Pathophysiology includes reduced urinary concentration capacity and loss of sodium conservation leading inevitably to renal failure.
Histological findings include uniformal thinning of renal cortex and a segmental distribution of cysts of varying sizes in the medulla and corticomedullary junction. The presence of cysts is a late finding and may not be found on a biopsy.
Clinical Presentation and Symptoms of MCK
Patients present with polyuria and polydipsia due to a reduced urinary concentration capacity. Median onset age for type 1 disease is 62 years and for type 2 is 32 years. Extrarenal manifestations are limited to hyperuricemia and gout. Anemia is common and may present before renal disease.
Laboratory Evaluation and Diagnosis of MCK
Urinalysis can be helpful and shows low specific gravity. Proteinuria is usually mild. The diagnostic study of choice is thin-section CT with contrast. It will show cysts in the medulla and corticomedullary junction. Ultrasonography is also helpful and will show normal or a moderate reduction in size with a loss of corticomedullary differentiation.
Therapy and Treatment of MCK
End stage renal disease develops in all cases and management is symptomatic. Erythropoietinis the treatment of choice for associated anemia. Eventually, patients will require dialysis and renal transplants.
Prognosis for MCK
Eventually, all patients develop end stage renal disease.
Multicystic Dysplastic Kidney (MCDK)
Epidemiology of MCDK
MCDK is one of the most frequent congenital kidney disorders. Unilateral MCDK occurs in 1 of 4300 live births and cumulative incidence of unilateral and bilateral occurs in 1 of 3600 live births. Involvement of the left kidney occurs in 55% of cases, whereas, that of the right kidney, occurs in 45%.
Etiology and Pathophysiology of MCDK
MCDK is mostly a sporadic disease, although familial inheritance has been reported. It is due to dysfunction in the early developmental period and is due to an abnormal induction of the metanephric mesenchyme (which later becomes kidney tissue) with the ureteral bud. Both exposure to viral infections, as well as spontaneous genetic mutations, have been shown to be involved in the pathogenesis.
MCDK may progress with no change in kidney size, may increase in size or may also end with involution. Calcification is a common feature that develops in adulthood, but can happen as early as 3 months after birth.
Histological examination usually reveals abnormalities in ductal differentiation and minimal corticomedullary differentiation. The kidney is usually large in size and resembles a collection of grapes.
Clinical Presentation and Symptoms of MCDK
At the present time, the disease is detected before birth and as early as 15 weeks of gestation with ultrasound. Before the advent of ultrasonography, newborns presented with a flank mass that is mobile, irregular in shape, non-tender and that could transilluminate.
Cases that are not detected on fetal ultrasound can present with urinary tract infections, hypertensions or dysfunction with voiding. They can also be found incidentally on imaging. MCKD is usually asymptomatic.
Laboratory Evaluation and Diagnosis of MCDK
Ultrasound is recommended as the preliminary diagnostic study. It shows a random arrangement of cysts of variable size and the renal pelvis cannot be identified. An annual blood pressure measurement is also recommended. Patients also should have ultrasound every 6 to 12-months until involution of the affected kidney.
Therapy and Treatment of MCDK
Nephrectomy is only performed for symptomatic patients and patients with complications. Those include abdominal or flank pain, urinary tract infections, hypertension and renal cancer.
Prognosis for MCDK
Prognosis is favorable and most cases of unilateral MCKD will have low morbidity and mortality. Morbidity is mostly caused by complications such as urinary tract infections, hypertension or neoplasia.
Acquired Cystic Kidney Disease (ACKD)
Definition and Epidemiology of ACKD
ACKD is defined by the development of numerous cysts without prior history of hereditary renal cystic diseases. It usually predates end stage renal failure and is commonly found incidentally in asymptomatic individuals. It has a significant association with dialysis and renal cell carcinoma. Other risk factors include male gender and African American ethnicity.
Etiology and Pathophysiology of ACKD
It is thought that uremia is the primary cause of ACKD. The association with dialysis is due to the fact that it prolongs the patient’s survival. Nonetheless, the exact mechanism remains unknown but is thought to involve ischemia and compensatory growth.
Presentation of ACKD
ACKD is asymptomatic in the early stages. It is present in every type of renal disease that causes a progressive loss of kidney function. Kidney involvement is usually bilateral and kidneys rarely become palpable.
Diagnosis and Laboratory Evaluation of ACKD
Ultrasonography is the diagnostic modality of choice. It will typically reveal no increase in renal size, as well as normal parenchyma. On histology, cysts are seen in the cortex, as well as in the medulla with deposition of oxalate crystals.
Therapy and Treatment of ACKD
There are no specific treatment recommendations for ACKD. Treatment is directed against loss of kidney function, as well as complications such as renal cell carcinoma, cystic infections and bleeding episodes.
Prognosis for ACKD
ACKD may result in significant complications. These include cystic infections, hemorrhage, erythrocytosis and malignant transformation.
Simple and Complex Cysts
Background and Epidemiology
Imaging techniques have become preponderant and, as such, asymptomatic cysts are routinely found. These cysts can be classified based on their radiologic characteristics and require different treatment. Simple renal cysts are far more common and their incidence increases linearly with age, with a prevalence of more than 30% in elderly individuals (> 70 year old). Complex cysts are associated with renal cell carcinoma.
Etiology and Pathophysiology
Causes for simple renal cysts remain unknown. One popular hypothesis postulates that they start forming from diverticula in the collecting tubules and collecting ducts. Simple cysts can also be present at birth, although occurrence between birth and the age of 20 is very rare. They are typically cortical and extend beyond the parenchyma and are filled with a homogeneous transudate. Studies have shown that they tend to increase very slowly in size.
Complex cysts, on the other hand, have distinguishing characteristics. They tend to show septae, calcifications, loculation and thickening of the walls. They also show increased density after imaging with contrast.
Diagnosis and Laboratory Evaluation
Simple or complex renal cysts are typically found incidentally on imaging, usually with CT scanning. A MRI can give a better resolution if further investigation is required to determine the stage of a complex cyst.

Image: “An arterial venous malformation of the left kidney and a simple cyst of the right kidney” by James Heilman, MD. License: CC BY-SA 3.0
Treatment and Prognosis
Simply cysts usually require no follow-up unless they are symptomatic or enlarging. Complex cysts, on the other hand, may require follow-up or surgical intervention due to the risk of renal cell carcinoma.
Review Questions
The answers are below the references.
1. What is the most frequent genetic determinant of autosomal dominant polycystic kidney disease?
- PKD1 on Chromosome 4
- PKD2 on Chromosome 4
- PKD1 on Chromosome 16
- PKD2 on Chromosome 16
2. Which cystic renal disease is associated with the most significant hepatic pathology?
- Autosomal Dominant Polycystic Kidney Disease
- Autosomal Recessive Polycystic Kidney Disease
- Medullary Cystic Disease
- Acquired Kidney Cystic Disease
- Complex cysts
3. What is a distinguishing extrarenal manifestation of medullary cystic kidney disease?
- Hyperuricemia
- Hepatic cysts
- Berry aneurysms
- No extrarenal manifestations with medullary cystic kidney disease
- Hypercalciuria



Gastric inhibitory polypeptide (GIP) is an important metabolic hormone in animals. It has a special molecular structure and plays an important physiological role in animal organisms. Gastric Inhibitory Polypeptide
ResponderEliminarThank you for the sharing of information wish you and your family a happy weekend ;)
EliminarThe herpes simplex virus is a contagious virus
ResponderEliminarthat can be
passed from person to person through direct
contact.
Children will often contract HSV-1 from early
contact with
an infected adult. They then carry the virus with
them for
the rest of their life.
Infection with HSV-1 can happen from general
interactions
such as eating from the same utensils, sharing
lip balm, or
kissing. The virus spreads more quickly when an
infected
person is experiencing an outbreak. Additionally,
it is
possible to get genital herpes from HSV-1 if the
individual
has had cold sores and performed sexual
activities during
that time.
HSV-2 is contracted through forms of sexual
contact with a
person who has HSV-2. It is estimated that around 20
percent of sexually active adults within the
United States
have been infected with HSV-2, according to the
American
Academy of Dermatology (AAD). ( AAD ) While
HSV-2
infections are spread by coming into contact
with a herpes
sore, the AAD reports that most people get
HSV-1 from an
infected person who is asymptomatic, or does
not have
sores.
having multiple sex partners
being female
having another sexually transmitted infection
(STI)
having a weakened immune system
TREATMENT
Herpes is a serious and recurring disease which can't be cured through drugs or injections by the American doctors but the best way to deal with Herpes is by taking natural herbs medicine for it, I have read about DR JAMES the great herbalist doctor who cured me from herpes with his powerful herbal medicine. I contacted him to know how he can help me and he told me never to worry that he will help me with the natural herbal mix after 2 days of contacting him, he told me that the cure has been ready and he sent it to me via UPS SPEED POST and it got to me after 3 days!i used the medicine as he instructed me (MORNING and EVENING) and I was cured!its really like a dream but I am so happy! for the people suffering from the following diseases, Cancer, hypothyroidism, Herpes, COPD, HIV, Arthritis, Hpv, Infections,Liver Disease, Autoimmune Diseases, Parkinson's disease,Lupus,Nephrology and Hypertension, Neurology, Obstetrics, Gynecology, and Women's Health, Oncology, Pediatrics, Pulmonary, ACUTE MYELOFIBROSIS, ALZHEIMER's symptoms, BREAST CANCER, DIABETES, HAIR LOSS AND HAIR TREATMENT, KIDNEY DISEASES, LEUKEMIA, MYELOID LEUKEMIA, STEM CELL TREATMENTand more should contact him for his herbal medicine because I am a living testimony and i was cured of herpes and his medicine is legit. I sent him what he requested and he sent me his medicine which I took for 3 good weeks and today am out here with negative result. when I went for test I was so happy after going through his medication. you can reach him through Whatsapp or call Him on +2348152855846 E-mail drjamesherbalmix@gmail.com