Proximal Convoluted Tubule (PCT): Clinical Effects of Fanconi Syndrome and Hartnup Disease
Table of Contents
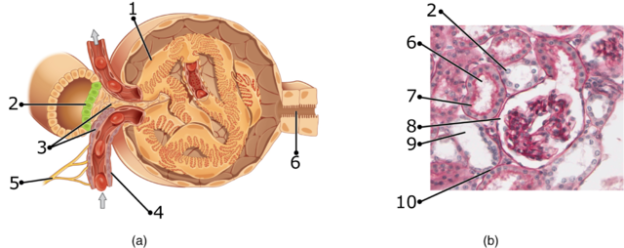
Image: “Juxtaglomerular Apparatus and Glomerulus numbers” by OpenStax College. License: CC BY-SA 3.0
Overview of PCT
The main characteristic and important feature of the proximal convoluted tubule is its apical brush border or striated border. The luminal surface of the tubule is densely covered with microvilli to increase the absorptive capability and surface area. It is easily visible under the light microscope and hence is given the name brush border. The removal of 80% of filtered urine is processed in PCT.
Ion-transport
The cytoplasm of the apical brush border cells is densely packed with mitochondria which gives them their acidophilic appearance. The mitochondria are present in large amounts in the basal region of the cells. The mitochondria are needed to provide energy for the active transport of ions. Sodium ions are transported out of the cells which creates an electrochemical gradient for more sodium ions to enter the cell from the luminal side followed by passive movement of water through tight junctions.
Cuboidal cells lining the proximal tubule interdigitate with one another along the lateral margins which makes it difficult to appreciate individual cells under the light microscope.
Divisions of PCT
The proximal tubule is divided into two sections:
- Pars convoluta (has higher cell complexity)
- Pars recta (has lower cell complexity)
Morphologically the convoluted segments of the proximal tubule are confined only to the renal cortex and straight segments go to the outer medulla.
Functions of PCT
1. Absorption of PCT
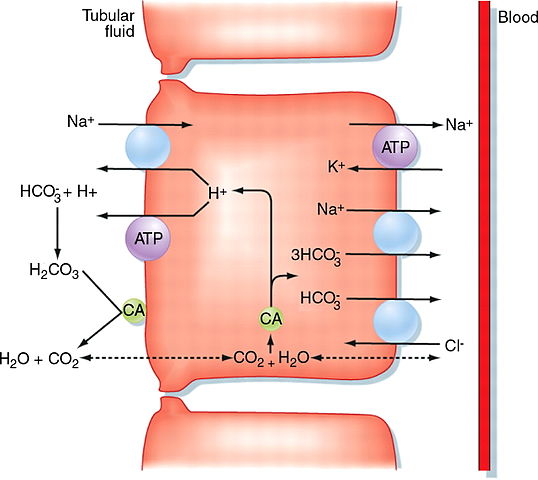
Image: “Proximal convoluted tubule” by M. Koeppen. License: CC BY-SA 4.0
The main function of the proximal convoluted tubule is absorption. It maintains and balances the pH by secreting hydrogen ions and absorbing bicarbonate ions. It secretes hydrogen ions with the help of pH Na- H antiporter in the luminal membrane.
Hydrogen ions which are secreted in combination with the bicarbonate ions to form carbonic acid. Na+ – K+ ATPase is present on the basolateral membrane of the epithelial cells which is responsible for transporting sodium from the luminal side into the blood. P-type ATPase is responsible for sodium reabsorption.
Substances mainly reabsorbed in the proximal convoluted tubule
- Salt and water: approximately two-thirds of the salt and water is reabsorbed by the transcellular transport followed by active reabsorption of sodium and passive absorption of water across the basolateral membrane by the sodium-potassium ATPase pump. 80 % of oxygen used by the kidney is required for the reabsorption of sodium into the blood.
- Organic solutes (glucose and amino acids): 100 % of glucose is reabsorbed by the secondary active transport through co transporters driven by the sodium gradient. PCT is the only place in the renal tubules where glucose is absorbed.
- Potassium: about 65 % of potassium is reabsorbed by two mechanisms which are solvent drag and simple diffusion.
- Urea: approximately 50 % of urea is reabsorbed via the solvent drag mechanism triggered by active sodium transport.
2. Secretion of PCT
The proximal convoluted tubule mainly secretes ammonium ions which are formed by the breakdown of glutamine to alpha-ketoglutarate. The proximal convoluted tubule also secretes many types of medications.
Clinical significance of PCT
Proximal tubular epithelial cells have a major role in kidney diseases. Most of the renal cell carcinomas arise in this portion of the nephron. Acute tubular necrosis results when proximal tubular epithelial cells are damaged by toxins, pigments, and sepsis due to reduced renal blood flow.
Overview of Fanconi Syndrome
Fanconi syndrome is a rare disease of the proximal renal tubule in which the proximal renal tubular cells are unable to absorb glucose, amino acids, uric acid, phosphate, and bicarbonate. It can be acquired or can be congenital. There are different forms of Fanconi syndrome which affects various functions of the proximal convoluted tubule. Renal tubular acidosis type 2 results when the reabsorption of bicarbonate is defective. Rickets and osteomalacia results when phosphate is not being reabsorbed.
Image: “Two-tier protection against genotoxic aldehydes” by Rationabilis. License: CC BY-SA 4.0
Fanconi syndrome has congenital or acquired causes. The acquired causes are exposure to chemicals or heavy metals, certain drugs, deficiency of Vitamin D, amyloidosis, kidney transplantation and multiple myeloma.
Pathophysiology of Fanconi Syndrome
There are numerous mechanisms which postulate the defective functioning of the proximal tubules resulting in the loss of substances but the three main mechanisms are:
- When the carriers responsible for transport of substances become defective.
- Alterations is cellular energy metabolism.
- When the permeability of the tubular membranes is altered.
There are numerous transporters located in the membrane of the proximal tubules which transport different substances across the cell. The energy required for this mechanism to take place comes from the sodium-potassium ATPase located in the basolateral membrane.
Many substances reduce the amount of ATP leading to Fanconi syndrome. One of these substances is maleic acid. Rats and dogs who were injected with maleic acid had a decrease in a number of sodium-potassium ATPase and decreased ATP resulting in glycosuria, phosphaturia, proteinuria, and bicarbonaturia. Similar results were observed when animals were injected with metals such as mercury, lead, and cadmium.
Most common cause of Fanconi syndrome in children is cystinosis which is caused by the accumulation of cystine in renal tubular cells. The genetic cause was postulated by a study in which five generation black family with autosomal dominant Fanconi’s syndrome was studied. The study postulated that mistargeting the peroxisomal EHHADH disrupts mitochondrial metabolism which leads to Fanconi syndrome. Abnormal accumulation of cysteine can induce an enlarged liver, eye disorders, and hypothyroidism.
Mortality and morbidity
The mortality is basically related to secondary causes caused by metabolic disturbances. Most of the secondary causes lead to developmental failures and renal failure.
Age, Race, and sex
Cystinosis only occurs in whites. The disease equally affects males and females.
Clinical features of Fanconi Syndrome
- Developmental delay
- Slow growth
- Polyuria
- Polydipsia
- Dehydration
- Osteomalacia in adults
- Hypophosphatemic rickets in children
- Acidosis
- Growth failure
- Hyperchloremia
- Hypokalemia
- Glycosuria
- Aminoaciduria
- Hyperuricosuria
- Fatigue
- Pain in the bones in adults
Diagnosis of Fanconi Syndrome
Urine analysis can be done which documents the loss of substances like amino acids, glucose, phosphate, and bicarbonate. Increased urinary lactate to creatinine ratio has recently been introducing which is a sensitive marker for the disordered proximal tubular function. Elevated levels of urinary retinol-binding protein 4 is a helpful biomarker helping in the diagnosis of Fanconi syndrome.
Treatment of Fanconi Syndrome
Treatment of patients with Fanconi syndrome mainly consists of replacing the substances like fluid and bicarbonate which are being lost in urine. Kidney transplantation can be considered in the cases of patients with renal failure caused due to cystinosis.
Overview of Hartnup Disease
Hartnup disease, also known as pellagra like dermatosis is an autosomal recessive disorder caused by defective absorption of non-polar amino acids particularly tryptophan which is further converted into melatonin, serotonin, and niacin. This disorder is characterized by defective transportation of neutral amino acids in both apical brush border of the small intestine and proximal tubule. Nicotinamide is formed by niacin which and nicotinamide is a component of NAD+ which is required for many metabolic functions.
Nicotinamide is required for neutral amino acid transporter production in proximal renal tubule in the kidney and in the intestinal mucosal cells in the small intestine. The defective gene is SLC6A19 which is located on chromosome 5. The patient represents distinctive skin rash and followed by uncoordinated voluntary movements, vision ailments, and cognitive delays.
Frequency
Overall 1 case in 30,000 live births has been found in screening programs done in Australia and North America. Overall prevalence has been reported as 1 case per 23000 live births which makes it the most common amino acid disorder in humans. Its manifestations start in the childhood.
Signs and symptoms
Signs and symptoms of hartnup disease usually become evident during infancy but sometimes begin as late as early adulthood. Symptoms can be triggered by fever, sunlight or emotional distress and can be summarized as the following:
- Failure to thrive
- Intermittent ataxia
- Nystagmus
- Photosensitivity (with rash on exposed parts)
- Tremor
- Pellagra (with associated diarrhea, dermatitis and dementia)
- Mental retardation
- Headaches
- Fainting
Pathophysiology of Hartnup Disease
Hartnup disease is an autosomal recessive disease and does not occur in heterozygotes. The disease results from cousin marriages or marriages within the same families for generations. The disease was reported in 1960 from the presence of indoles and tryptophan in the urine of the affected individuals. Pellagra-like symptoms were caused by excessive loss of tryptophan due to malabsorption. In 2004 the causative gene was identified which was SLC6A19 which was located on chromosome number 5.
It is a sodium-dependent and chloride independent transporter which transports neutral amino acids like tryptophan and is expressed in large amounts on the kidneys and intestine.
Diagnosis of Hartnup Disease
Patients with the hartnup disease are unable to absorb tryptophan which is required for many metabolic functions. Thus, in times of stress or need, their body is unable to make an adequate amount of B-complex vitamin niacinamide.
For the diagnosis of hartnup disease, urine chromatography is done which shows increased urinary excretion of the following neutral amino acids:
- Glutamine
- Valine
- Phenylalanine
- Leucine
- Citrulline
- Asparagine
- Alanine
- Serine
- Tyrosine
- Tryptophan
- Indican (can also be tested by Obermayer test)
Skin biopsy and Jejunal biopsy is done in selected patients. Skin changes are similar to the skin changes in pellagra.
Molecular genetic testing provides the confirmation of the Hartnup disease by detecting variations in SLC19A6 gene.
Treatment of Hartnup Disease
Treatment aims at avoiding exposure to the things which exaggerate or trigger an attack and aiming at replenishing the deficient amino acids.
Following are the treatment modalities used:
- High protein diet
- Use of physical or chemical protection from sunlight by wearing caps and using high SPF sunscreens
- Avoidance of photosensitizing drugs
- Daily supplementation with nicotinic acid in niacin deficient patients
Prognosis of Hartnup Disease
Most of the patients affected remain asymptomatic but in some patients, skin manifestations and neurologic involvement really impair quality of life. Attacks become less frequent with age. Mental retardation and short stature is seen in some of the patients Pregnancy is not influenced by hartnup disease as the placental transport of free amino acids is not being affected.
Overview of TYPE 2 (Proximal) Renal Tubular Acidosis
Proximal renal tubular acidosis is caused by the failure of the proximal tubular cells to reabsorb the filtered bicarbonate which leads to bicarbonate loss. As bicarbonate is a major buffer in the body, its loss leads to relative acidemia. The distal intercalated cells are unaffected and function normally so the academia is much less severe than the distal renal tubular acidosis.
There are many causes of proximal renal tubular acidosis but one of the major known causes is Fanconi syndrome. Due to secondary hyperaldosteronism, the patients are also typically hypokalemic and there is urinary potassium loss as well.
Causes of TYPE 2 (Proximal) Renal Tubular Acidosis
There are acquired and familial causes leading to proximal renal tubular acidosis which are listed below.
Acquired disorders
- Multiple myeloma
- Paroxysmal nocturnal hemoglobinuria
- Amyloidosis
- Heavy metals like lead and cadmium
- Familial disorders
- Glycogen storage disease type 1
- Galactosemia
- Cystinosis
- Tyrosinemia
- Wilson’s disease
- Lowe syndrome
- Secondary hyperparathyroidism
Treatment of TYPE 2 (Proximal) Renal Tubular Acidosis
Treatment mainly consists of oral bicarbonate supplementation. Potassium supplementation should always be done prior to bicarbonate supplementation otherwise it can lead to worsened hypokalemia. Oral bicarbonate administration can lead to bicarbonate diuresis thus the amount of bicarbonate given should be large keeping urinarily loses in mind. Thiazide diuretics can also be given to the patients. Vitamin D and calcium supplements are given to reduce skeletal deformities represented from osteomalacia or rickets.



{kind=link}
{kind=link}
.jpg){kind=link}
Comentários
Enviar um comentário