Microcytic Anemia: Thalassemia (Alpha and Beta Thalassemia)
Table of Contents
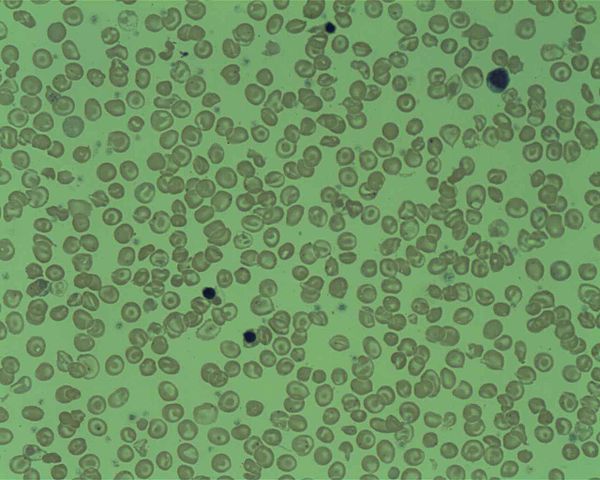
Image: “Peripheral Blood film from a patient with Delta Beta Thalassemia.” by Prof. Osaro Erhabor. License: CC BY-SA 4.0
Definition of Thalassemia
Thalassemia is a microcytic-hypochromic anemia. Its cause is a decreased synthesis of one or several globin chains. Since globin synthesis is flawed, the disease is one of the so-called hemoglobinopathies. Depending on which globin chain is affected by the disorder, one speaks of α- or ß-thalassemia.
Thalassemias are caused by gene mutations leading to decreased production of globin protein. Deletions of α-globin genes cause α-thalassemia. There are two α-globin genes closely linked on chromosome 16. There is a total of 4 alleles.

Epidemiology of Thalassemia
Since thalassemia more frequently occurs in the Mediterranean area, including Turkey, it is also referred to as ‘Mediterranean anemia.’ However, it can be found worldwide. It is estimated that ca. 3% of the world population carry at least one ß-thalassemia gene. In Germany, the most frequent form is heterozygous ß-thalassemia.
Etiology of Thalassemia
Thalassemia is an inherited autosomal-codominantly. At heterozygosity, a milder form of the disease develops (minor thalassemia). At homozygosity, the severe form (major thalassemia) can be observed. There is also an intermediate thalassemia form. The primary causes are genetic defects, which form the pathogenetic basis of the decreased synthesis of one of several polypeptide chains of the globin molecule.
In turn, this results in a decreased hemoglobinization of the erythroblasts. In the blood count, this condition can be observed as hypochromasia of the erythrocytes. Additionally, the globin chains, which continue being produced, aggregate and make for increased apoptosis of precursor cells via different pathophysiological mechanisms. This condition also leads to decreased lifespan of the erythrocytes in the peripheral blood.
α-Thalassemia
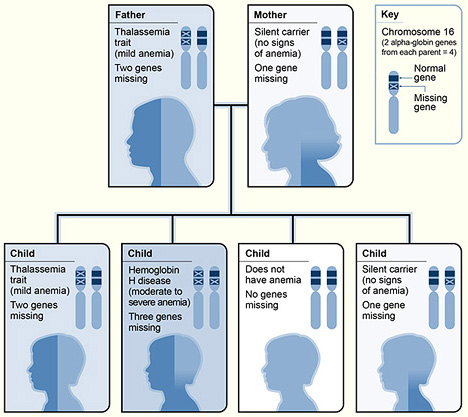
The pictograph shows one example of how alpha thalassemia is inherited. The alpha globin genes are located on chromosome 16. A child inherits four alpha globin genes (two from each parent). In this example, the father is missing two alpha globin genes and the mother is missing one alpha globin gene. Each child has a 25 percent chance of inheriting two missing genes and two normal genes (thalassemia trait), three missing genes and one normal gene (hemoglobin H disease), four normal genes (no anemia), or one missing gene and three normal genes (silent carrier).
The characteristics and severity of the disease depend on the number of genes deleted:
Hemoglobin Electrophoresis

Newborns: Splenic macrophages phagocytose and destroy RBCs with HbF and replace them with HbA and HbA2, which takes a few months.
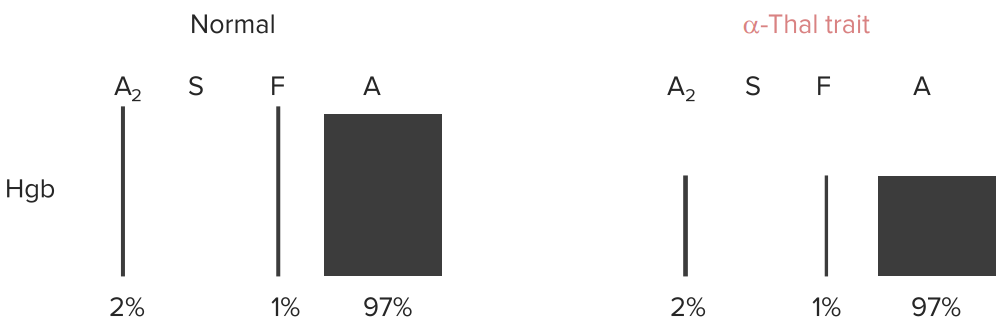
Anemia, but no change in % of Hb A, A2 or F (all of them need α-globin chains for their synthesis).
β-Thalassemia
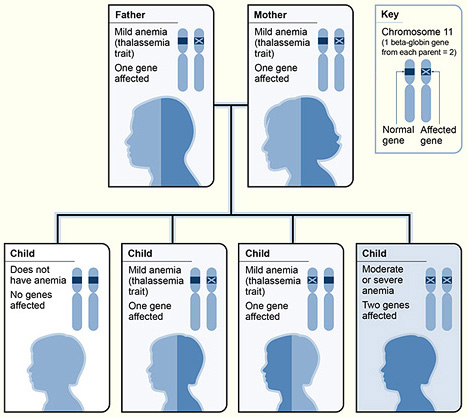
The pictograph shows one example of how beta thalassemia is inherited. The beta globin gene is located on chromosome 11. A child inherits two beta globin genes (one from each parent). In this example, each parent has one altered beta globin gene. Each child has a 25 percent chance of inheriting two normal genes (no anemia), a 50 percent chance of inheriting one altered gene and one normal gene (beta thalassemia trait), or a 25 percent chance of inheriting two altered genes (beta thalassemia major).
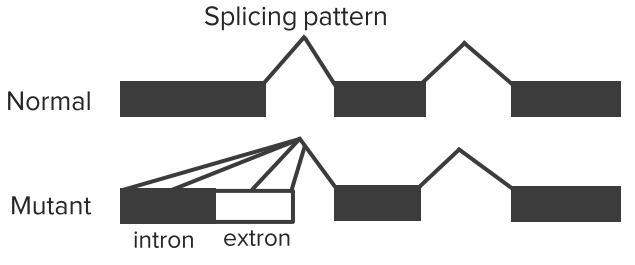
There is only one β-globin gene on chromosome 11, total of 2 alleles. β-Thalassemia is primarily caused by splicing mutations in β-globin genes.
There are two types of mutations in the β-globin gene:
- β+ mutations – variable decreased expression;
- β0 mutations – absent expression.
This results in three different clinical syndromes:
Clinic of Thalassemia
Depending on the genetic defect, thalassemia shows a variable clinical picture.
Minor thalassemia usually does not show severe symptoms. Slight hepatosplenomegaly and possibly a recurrent jaundice in a mild form can be present. Also, target cells can be observed in the blood count.
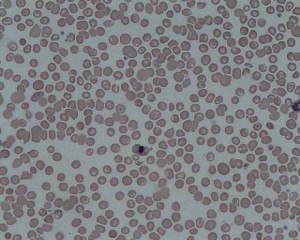
Image: “Target-Cells” by Osaretin. License: CC BY-SA 4.0
In its more distinct form, that is major thalassemia, the disease can lead to bone deformations, and even fractures, due to the reactive expansion of erythropoiesis in the bone marrow. Because of the decreased, and often pathological erythrocytes, hypoxia becomes clinically relevant as growth disturbances and trophic skin changes show in child age.
α-Thalassemia
There are two consequences of decreased α-globin production:
- Decreased hemoglobin synthesis, causing microcytic, hypochromic anemia;
- Quantitative imbalance between α- and β-globin proteins, resulting in the formation of insoluble β-globin (HbH) or γ-globin (Hb Barts) aggregates in the RBC. These RBCs are often cleared in the spleen and liver, worsening the anemia.
It is most common in patients of Southern Asian (–allele) and African (-α allele) descent.
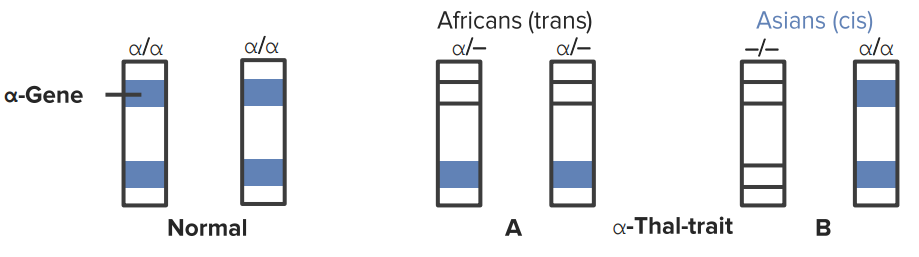
α-Globin chain gene deletions
2 deletions (trait)
3 deletions (HbH; 4 β chains)
4 deletions (Hb Bart´s; 4 γ chains)
HbH disease
- Signs and symptoms of anemia;
- Chronic hemolysis with variable jaundice and cholelithiasis (bilirubin stones);
- Extramedullary hematopoiesis with frontal bossing and hepatosplenomegaly.
Hydrops fetalis
- Anasarca (generalized edema) from high-output heart failure;
- Hepatosplenomegaly;
- Causes death in the prenatal period.
β-Thalassemia
Similar to α-Thalassemia.
- AR disorder: Africans, Italians, Greeks;
- → RBC count: splicing defect for minor; nonsense mutation for major (stop codon).
β = normal
β+ = some
β0 = none
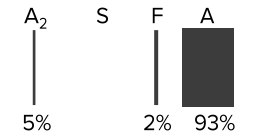
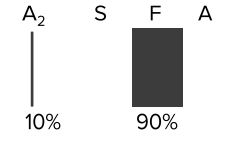
β-Thalassemia major (Cooley’s anemia)
Laboratory findings:
- Decreased Hgb, Hct and MCV;
- Variable RBC count;
- Hemoglobin electrophoresis varies depending on the severity of the disease, with decreased HbA, increased HbF (α2γ2) and increased HbA2 (α2δ2).
It causes severe transfusion dependent anemia that develops at a few months of age (as HbF declines). If adequately transfused, children will develop normally, but will develop secondary hemochromatosis and die of heart disease in their 20s. If not transfused, there is a stunted growth, bony changes and high-output heart failure with death in infancy.
Diagnosis of Thalassemia
The blood count of minor thalassemia shows microcytic, hypochromic erythrocytes. Since this is also the case with iron deficiency and this condition is more frequent in practice, one should consider minor thalassemia when confronted with a non-confirmed iron deficiency anemia. A blood smear with target cells and poikilocytosis provides additional certainty.
- Laboratory studies: decreased Hgb, Hct, MCV; increased RBC count, RDW; HbH on hemoglobin electrophoresis; iron studies normal.
At major thalassemia and intermediate thalassemia, hypochromasia and poikilocytosis are more distinct. Reticulocytes, LDH and bilirubin are increased; haptoglobin is decreased.
Further criteria confirming the suspicion are a positive family history, disturbed hemoglobinization in bone marrow aspirate and genome analysis.
Therapy of Thalassemia
The ideal therapy is allogenic stem cell transplantation. Since the minor form does not usually require treatment, this primarily applies to major thalassemia.
Course and Prognosis of Thalassemia
If the anemia is not treated adequately, there is a massive expansion of hemopoiesis within the bone marrow. This can lead to a “hair on end” appearance on skull X-ray. If regular transfusions are given, iron chelation must be started.
While minor and intermediate thalassemia mostly progress without complications, major thalassemia can lead to death at infant age without treatment. Another life-shortening factor is increased iron accumulation (hemosiderosis), which is why iron supplementation is contraindicated in cases of thalassemia.



{kind=link}
Thalassemia is a blood disorder passed down through families (inherited) in which the body
ResponderEliminarmakes an abnormal form or inadequate amount of hemoglobin.