Pediatric Biliary Atresia — Diagnosis and Surgery
Biliary atresia is an acquired obliteration of the biliary tree leading to neonatal cholestasis, secondary biliary cirrhosis, and portal hypertension. It is the most common indication for liver transplant in children. This article discusses in depth the clinical presentation, classification, etiopathogenesis, workup and management of biliary atresia.
Table of Contents
- Introduction
- Epidemiology of Pediatric Biliary Atresia
- Etiopathogenesis of Pediatric Biliary Atresia
- Classification of Pediatric Biliary Atresia
- Clinical Presentation of Pediatric Biliary Atresia
- Investigations of Pediatric Biliary Atresia
- Treatment of Pediatric Biliary Atresia
- Complications
- Prognosis
- Review Questions
- References
Are you more of a visual learner? Check out our online video lectures and start your pediatric gastroenterology course now for free!
Introduction
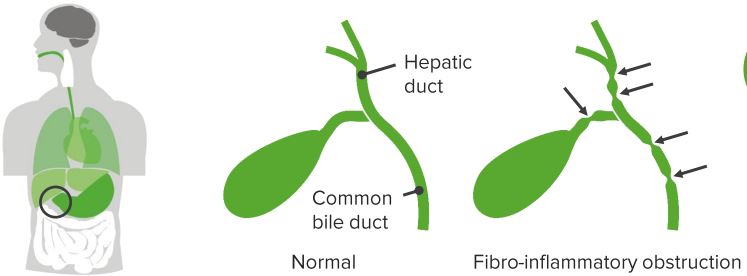
Biliary atresia is the obliteration of bile ducts. Extrahepatic ducts are much more profoundly affected than the intrahepatic ducts. The extent of involvement of bile ducts can be variable. Biliary atresia is the most common cause of neonatal cholestasis.Epidemiology of Pediatric Biliary Atresia
Biliary atresia occurs with a frequency of 1:8000 to 1:13000 live births. Incidence is almost equal among both genders but for a slight female predominance. It is not an inherited disorder (there is no increased risk among kindred or siblings). Biliary atresia shows significant seasonal clustering with incidence increasing substantially in winter months.Etiopathogenesis of Pediatric Biliary Atresia
The exact etiology of biliary atresia is unknown. There is a progressive injury to the epithelial lining of bile ducts. Some perinatal insult, possibly a viral infection, incites the injury and the subsequent inflammatory response propagates it. The bile duct lumen is filled with the inflammatory plug. Subsequent organization leads to fibrosis and obliteration of the bile ducts.Although extrahepatic ducts are mainly affected, the pathology also extends to intrahepatic ducts (but to a lesser extent). This coupled with backpressure changes due to the obliteration of extrahepatic ducts leads to liver injury and secondary biliary cirrhosis with portal hypertension.
If appropriate treatment is not instituted in time, liver cirrhosis and subsequent liver failure are often fatal within three years of age.
“Biliary Atresia” Image created by Lecturio
Classification of Pediatric Biliary Atresia
Depending on the timing of presentation, biliary atresia is subdivided into fetal/embryonic form and perinatal/postnatal form. The fetal form is less common (10 to 35% of cases). The infants are jaundiced from birth. The fetal form is believed to be due to defective embryogenesis with ductal plate malformations. This also accounts for increases in the risk of associated malformations like “polysplenia syndrome.”The perinatal or postnatal form is due to perinatal insult, possibly a viral infection which triggers the obliterative process leading to atresia. This is the more common variety. The infant is well at birth and progressively increasing jaundice starts a few weeks later.
Biliary atresia can be subdivided into three sub-groups depending on the extent of involvement of bile ducts. The same is detailed in the following table.
| Subtype | Morphology |
| Type I | Atresia restricted to common bile duct. |
| Type II | Atresia of common hepatic duct. |
| Type III | Atresia of right and left hepatic ducts (Intrahepatic involvement). |
Clinical Presentation of Pediatric Biliary Atresia
About one-third of the cases are jaundiced at birth with progressive increase thereafter (fetal subtype). The rest present with progressively increasing jaundice from 3 to 6 weeks of age. There is typically paucity of pruritus. A complete lack of bile pigments causes chalky white stools. These acholic stools can be intermittent or delayed in onset. Urine is typically high colored or cola colored. On examination, the child is icteric with hepatosplenomegaly. The liver is enlarged and is soft to firm. The spleen is enlarged in view of the accompanying portal hypertension.Investigations of Pediatric Biliary Atresia
Aspiration of duodenal contents
If there is no atresia of the bile ducts, the bile flows freely into the duodenum along its normal physiological path. Aspirating the duodenal contents after intubating the duodenum is a simple test to exclude biliary atresia. The presence of bile in the duodenal aspirate rules out biliary atresia; however, this test is invasive and uncomfortable to the patient.Hepatobiliary iminodiacetic acid scan (HIDA scan)
HIDA scan evaluates the function of the hepatocytes as well as the excretory pathway. With optimal hepatocyte function, the isotope is taken up by the liver and excreted into the biliary tree. The presence or absence of activity in the intestine is evaluated. In biliary atresia, the hepatocyte function is relatively well preserved. The liver shows a good uptake of the isotope, but there is no excretion into the intestine. In contrast to neonatal hepatitis, due to impaired hepatocyte function, the liver shows an impaired uptake but excretion into the intestine eventually occurs.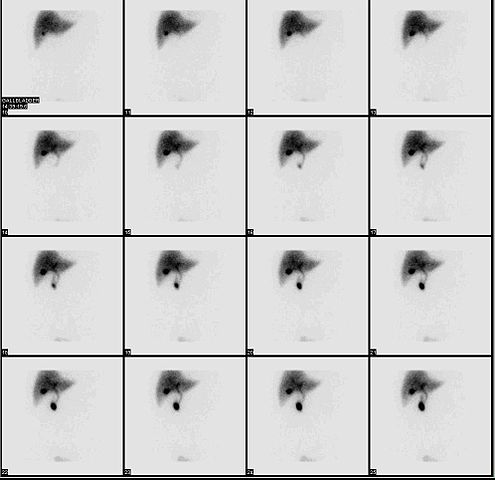
Pre-procedure administration of phenobarbitone increases the biliary excretion of the isotope, thus increasing the sensitivity of the test.
Ultrasound
It is a good non-invasive investigation which is widely available. Dilated cysts are identified in patients with a choledochal cyst. When the gallbladder is involved in biliary atresia, it is not visualized. “Triangular sign” is highly suggestive of biliary atresia. A fibrous cone of tissue at the bifurcation of the portal vein is visualized. Despite being a dynamic investigation, ultrasound is highly operator dependent.Liver biopsy
A percutaneous liver biopsy is useful, providing a definitive diagnosis in up to 95% of patients. Typically in patients with biliary atresia, there is the proliferation of bile ducts, stasis of bile at the canalicular and cellular level and edema and fibrosis in the perilobular area. The presence of bile plugs in the portal tracks is specific for biliary atresia but is found in about only 40% of specimens. Later in the course of the disease, extensive fibrosis consistent with developing secondary biliary cirrhosis can be seen. Giant cell transformation of the hepatocytes can be seen in both biliary atresia and neonatal hepatitis.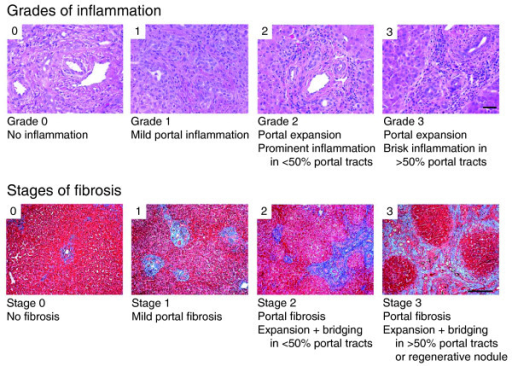
Image: “Representative photographs of portal tracts stained with hematoxylin/eosin (upper panel) used for grading of liver sections in biliary atresia based on the presence of inflammatory cells (scale bar on photo 3 = 50 μm). The lower panels depict liver sections stained with trichrome for staging based on the extent of fibrosis (scale bar on photo 3 = 250 μm).” By Moyer K, Kaimal V, Pacheco C, Mourya R, Xu H, Shivakumar P, Chakraborty R, Rao M, Magee JC, Bove K, Aronow BJ, Jegga AG, Bezerra JALicense: CC BY 2.0
Surgical exploration with on table cholangiography
When the clinical suspicion is high but the preoperative investigations are equivocal, there is a role of surgical exploration with intraoperative cholangiography (IOC). Assessment of extrahepatic bile ducts is done. Bile ducts are cannulated and intraoperative cholangiography was done. It helps to document the site of obstruction and direct the appropriate surgical therapy.Differential Diagnosis of Neonatal Cholestasis
Neonatal cholestasis can be caused by a large variety of disorders including:Biliary Cyst:
It’s considered a rare disorder, that can be diagnosed by ultrasonography, and mostly is managed surgically during neonatal period to prevent the development of ascending cholangitis.
Cystic fibrosis, with inspissated plug syndrome.
Gallstones with biliary obstruction.
Neonatal hepatitis, where the differentiation can sometimes be difficult.
A detailed clinical history and physical examination, combined with appropriate diagnostic studies, are usually able to differentiate between these. Differentiating neonatal hepatitis from biliary atresia is particularly difficult. Both cause giant cell transformation of the hepatocytes.
Treatment of Pediatric Biliary Atresia
The definitive treatment of biliary atresia remains surgery. Kasai hepatoportoenterostomy is the procedure of choice today.A small proportion of patients has “correctable” anatomy (Type I anatomy). If the distal bile duct is obliterated but there is a remnant proximal bile duct which is in continuity with intrahepatic bile ducts, a bilioenteric anastomosis can be fashioned at the level of hilum (Hilar hepaticojejunostomy) and adequate biliary decompression provided. In all other cases, Kasai hepatoportoenterostomy is the advocated procedure for initial management.
Kasai Hepatoportoenterostomy
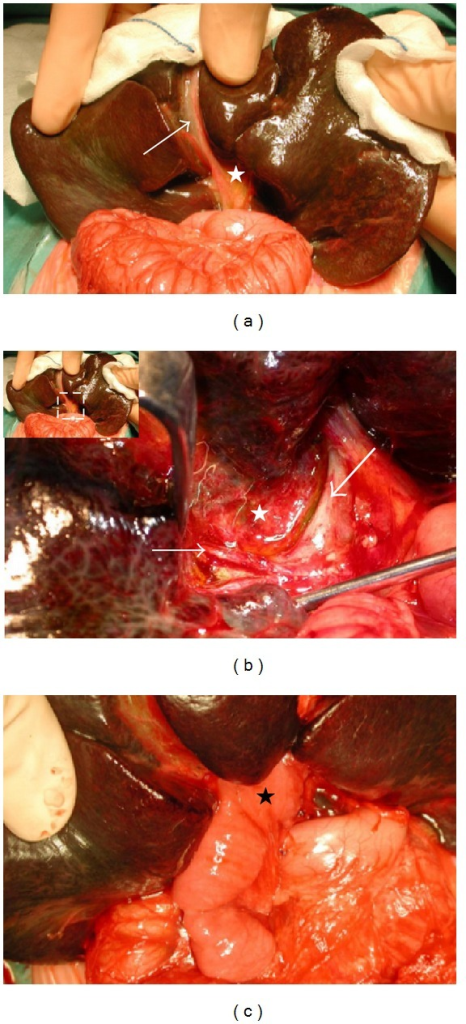
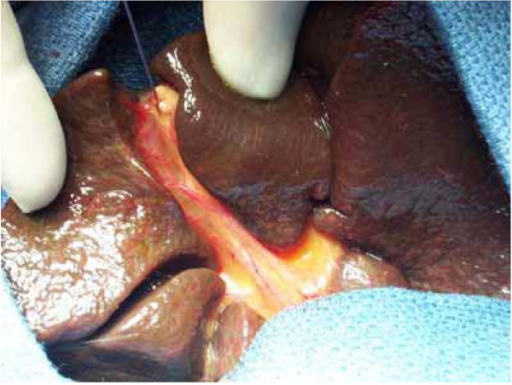
Image: “(a) Complete biliary atresia with a shrunken, fibroticgall bladder (arrow) and biliary remnant (white star) in a 2-month-old baby. Note the already cirrhotic aspect of the liver. (b) Hilar region after removal of the biliary remnant (white star), surrounded by the hepatic artery’s right branch (fine arrow) and the portal vein’s left branch (large arrow). (c) The final aspect of the Kasai hepato-porto-enterostomy with the Roux-en-Y limb (black star) anastomosed to the hilar region.” by Barbara E. Wildhaber. License: Open Access
The ductal plate at porta hepatis has microscopic bile ducts. When these ducts are in continuity with intrahepatic bile ducts, there is a reasonable possibility of adequate biliary decompression and resolution of jaundice. If the communication is absent or the size is very small, the procedure may fail.
The timing of surgery has a direct bearing on the postoperative outcomes. This is the reason why expedient diagnosis and institution of appropriate therapy is of paramount importance in biliary atresia. When infants are operated upon within 60 days of birth, the bile flow is re-established in about 80% of cases; however, this success rate falls dramatically as the delay increases. Infants more than 90 days of age have a poor 20% chance of re-establishment of bile flow.
The excised fibrous tissue at porta hepatis is sent for histopathological examination. The presence of microscopic ducts of 150 μm or more in diameter is predictive of good postoperative outcome. Other factors which influence the postoperative outcomes are a degree of proliferation of periductular glands, technical expertise of the operating surgeon and the development of postoperative complications. Complete resolution of jaundice at three months post procedure is an indicator of good long-term success.
Bacterial cholangitis is common in the postoperative period and many surgeons keep these patients on long-term oral antibiotic therapy at follow-up. Repeated episodes of bacterial cholangitis lead to the obliteration of microscopic ducts at the porta and a long-term failure of the procedure.
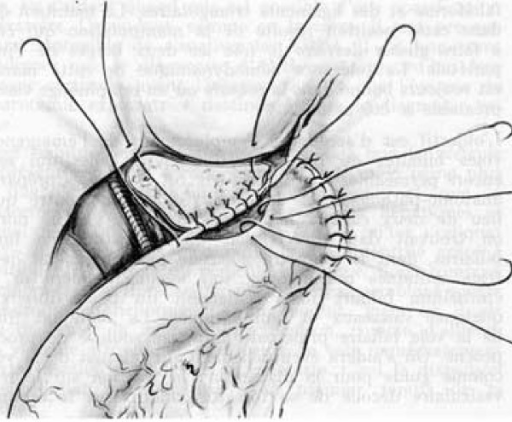
Re-operation Following Kasai Procedure
Occasionally, in a small sub-group of patients, post successful Kasai Hepatoportoenterostomy, bile drainage may abruptly stop. Redo surgery can be done in these. The fibrotic scarred area at porta is excised and redo portoenterostomy done. The same cannot be done in the patients who had poor bile flow after the index procedure.Liver Transplantation
Biliary atresia is the most common indication for liver transplantation in pediatric age group. Although results with Kasai’s procedure are good, eventually a liver transplant is required in up to 80% patients. With the improvement in surgical expertise and perioperative care, the results of liver transplant have consistently improved. The long-term outcomes post-transplant are good.Indications for liver transplantation in biliary atresia are failed hepatoportoenterostomy (liver decompensation, intractable portal hypertension and recurrent bleeds, and refractory growth failure). Anastomotic stenosis, recurrent cholangitis, progressive pathology with the loss of intrahepatic ducts contributes to the long-term failure of Kasai procedure.
Complications
Following portoenterostomy, children with biliary atresia can develop acute and chronic complications. During the early days after the operation, the most common complication isunsuccessful anastomosis which failed to achieve adequate bile drainage.Over time, in about 60% of children, there are complications with progressive liver disease and portal hypertension. Fifty percent of children eventually develop cholangitis after a portoenterostomy.
Patients who have cirrhosis without any evidence of portal hypertension are at risk of developing a hepatocellular carcinoma. For children who fail to drain out bile, they may develop progressive fibrosis and biliary cirrhosis. In such cases, liver transplant is the last treatment option.
Prognosis
For children who undergo a hepatoportoenterostomy when they are 70 days of age, administering medium to high-dose of steroid therapy produces a good outcome with regards to jaundice clearance. However, children need to undergo longer follow up routine to ensure that the effect of steroid treatment is positive.Review Questions
The correct answers can be found below the references.1. All the following is true about Biliary atresia except:
- It is the commonest indication for liver transplantation in the pediatric population.
- Diagnosed cases of biliary atresia were usually well at birth with normal birth weight.
- Children with biliary atresia commonly present with conjugated hyperbilirubinemia at five to ten years of age.
- The gallbladder may or may not be involved in the fibrotic process.
- Surgery was done at 4 weeks of age.
- Surgery is done by a senior surgeon with considerable past experience of managing similar cases.
- Surgery was done at 4 months of age.
- None of the above.
- Jane should be reassured that all is well and the yellowish discoloration of eyes will subside this time too.
- Jane should be counseled that this needs to be investigated further.
- Laboratory tests like serum total bilirubin, conjugated bilirubin, AST, ALT, Alkaline phosphatase will help in diagnosis.
- Diagnosis should be established as soon as possible and appropriate treatment instituted without any further delay.
- Aspiration of bile stained fluid from duodenum rules out biliary atresia.
- A well conducted triple phase contrast enhanced CT scan of the abdomen can diagnose the underlying cause of cholestasis in 99 percent of affected children.
- On ultrasound, a triangular cord sign is seen in biliary atresia, while dilation of the bile ducts is seen in the choledochal cyst.
- A liver biopsy can help in the diagnosis.
- Liver transplantation is advocated for children who fail Kasai’s hepatoportoenterostomy.
- Once failed, a repeat hepatoportoenterostomy should never be attempted.
- Success after Kasai’s hepatoportoenterostomy is influenced by anastomotic stenosis, ascending cholangitis and status of loss of intrahepatic bile ducts.
- In the fetal form of biliary atresia, the infant is jaundiced from birth with no jaundice free interval



{kind=link}
Comentários
Enviar um comentário