Marfan Syndrome (MFS) — Signs and Symptoms
Table of Contents
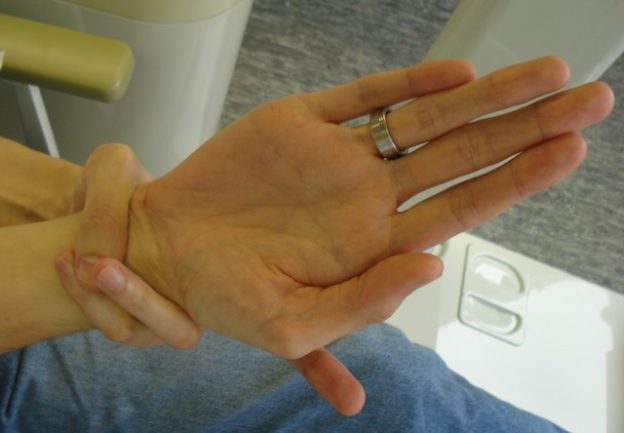
Image: “A positive wrist sign in a patient with Marfan syndrome. In case of a positive wrist sign the thumb and little finger overlap, when grasping the wrist of the opposite hand.” by Staufenbiel I, Hauschild C, Kahl-Nieke B, Vahle-Hinz E, von Kodolitsch Y, Berner M, Bauss O, Geurtsen W, Rahman A – http://www.ncbi.nlm.nih.gov/pubmed/24165013. Licence: CC BY-SA 2.0
Definition of the Marfan Syndrome
Marfan syndrome (MFS) was first described in 1896 by Antoine Marfan, a pediatrician in Paris. Unlike many other diseases, there is equal distribution between the genders and prevalence in all parts of the world. MFS is inherited in an autosomal dominant pattern or caused by spontaneous mutation and is about as frequent as mucoviscidosis.
The mutation is located on chromosome 15q21 (Marfan locus). This long arm of the chromosome 15 includes the gene encoding for fibrillin-1. This leads to a disturbed synthesis of microfibrils and thus elastic fibers.
Pathology of the Marfan Syndrome
Marfan syndrome is an autosomal dominant disorder of the connective tissue. It affects multiple systems, e.g. the cardiovascular and skeletal systems and the eyes. It occurs in 1/5000 live births.
Marfan Traits
- Pes planus
- Crowded teeth
- Tall, thin stature
- Elongated extremities
- Flexible joints
- Pectus excavatum/carinatum
- Scoliosis
Three body systems are mainly affected:

Lens dislocation in Marfan syndrome with the lens being kidney-shaped and resting against the ciliary body
Ocular system (eyes): Ectopia lentis, retinal detachment.
Musculoskeletal system: dolichostenomelia (decreased upper: lower segment ratio), Arachnodactyly, thoracic cage deformities.
Cardiovascular system (heart/aorta): aortic dissection, mitral valve insufficiency.
The degree of the manifestations varies to a high extent! The main cause of death, aortic dissection, is, however, often not detected. Typical manifestations of neonatal MFS are, in addition to those mentioned above, dolichocephaly, malar hypoplasia, enophthalmos, retrognathia and an impaired range of motion of peripheral joints.
Genetics of the Marfan syndrome
- Mutation in FBN 1 gene on chromosomes which encodes fibrillin-1
- 75% of patients with MFS have affected parents
- 25% de novo mutation
Skeletal abnormalities
- Tall stature
- Long headed
- Arachnodactyly (long thin digits)
- Hyperextensible joints
- High arched palate
- Scoliosis, kyphosis, slipping off vertebrae
- Pectus excavatum
Ocular changes
- Ectopia lentis: Dislocation of the lenses
- Myopi
- Retinal detachment
- Early glaucoma
- Early cataract
Cardiovascular lesions
- Cardiovascular symptoms are main cause of morbidity and mortality
- Degeneration of the tunica media
- Dilated, incompetent aortic root with valvular incompetence
- Mitral valve prolapse and regurgitation
- Aortic aneurysms may dissect or rupture
- Monitoring by echocardiography is required!
Neurological leasions
60% of the patients who have Marfan syndrome are known to have dural ectasia.
Diagnosis of the Marfan Syndrome and Ghent Nosology
Ghent nosology was introduced in 1996 and has made a diagnosis of MFS easier worldwide by resuming all diagnostic criteria. If the family history is negative, three major criteria in two different body systems must be present and a third system has to be involved. It was revised in 2010. There are seven new criteria that lead to a diagnosis.
In the absence of a family history of MFS:
- Aortic root Z-score ≥ 2 AND ectopia lentis
- Aortic root Z-score ≥ 2 AND an FBN1 mutation
- Aortic root Z-score ≥ 2 AND a systemic score > 7 points
- Ectopia lentis AND FBN1 mutation with known aortic pathology
In the presence of a family history of MFS:
- Ectopia lentis
- Systemic score ≥ 7
- Aortic root Z-score ≥ 2
Points for systemic score:
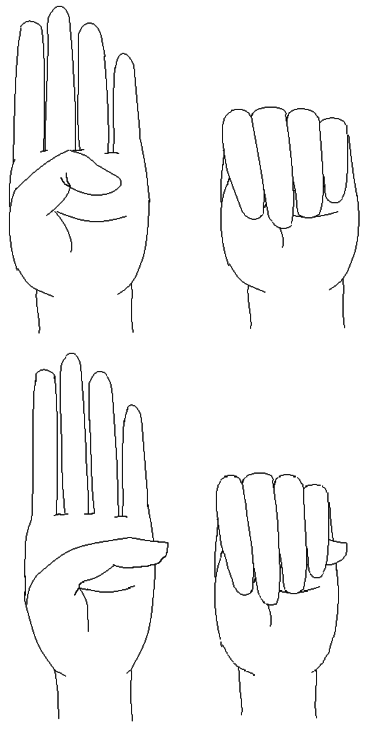 Image: “Steinberg’s thumb sign (Marfan’s syndrome) – a flexed thumb grasped within a clenched palm protrudes beyond the ulnar border of that hand.” by Goopsmirk – Own work. Licence: CC BY-SA 3.0Wrist AND thumb sign = 3 (wrist OR thumb sign = 1)
Image: “Steinberg’s thumb sign (Marfan’s syndrome) – a flexed thumb grasped within a clenched palm protrudes beyond the ulnar border of that hand.” by Goopsmirk – Own work. Licence: CC BY-SA 3.0Wrist AND thumb sign = 3 (wrist OR thumb sign = 1)- Pectus carinatum deformity = 2 (pectus excavatum or chest asymmetry = 1)
- Hindfoot deformity = 2 (plain pes planus = 1)
- Dural ectasia = 2
- Protrusio acetabuli = 2
- Pneumothorax = 2
- Reduced upper segment/lower segment ratio AND increased arm/height AND no severe scoliosis = 1
- Scoliosis or thoracolumbar kyphosis = 1
- Reduced elbow extension = 1
- Facial features (3/5) = 1 (dolichocephaly, enophthalmos, down-slanting palpebral fissures, malar hypoplasia, retrognathia)
- Skin striae (stretch marks) = 1
- Myopia > 3 diopters = 1
- Mitral valve prolapse ¼ 1

A systemic participation is available when 7 or more points are reached.
Treatment of Marfan Syndrome
Patients can only be treated individually for symptomatic management, but the most important part is to have regular appointments with ophthalmologists, cardiologists and orthopedic specialists, as they treat the systems that are mainly affected. It is crucial to monitor the aortic root diameter being assessed echocardiographically. Prospective and retrospective trials have shown that propanol and atenolol can slow down aortic dilatation, resulting in an increased usage of beta blockers. This also helps prevent or forgo necessary cardiac surgery.
Life expectancy for patients with MFS has increased in the past few years. This is mainly attributed to cardiac surgery that patients undergo soon after the diagnosis to prevent rupture and dissection of the aorta.
Multidisciplinary Therapy
- Cardiology, genetics, ophthalmology, orthopedics, cardiothoracic surgery
- Follow with echo
- Beta-blockers
- Angiotensin II inhibitors/transforming growth factor-beta inhibitors
- Strict blood pressure control
- Aortic graft for aortic dilation > 5 cm
- Avoid caffeine, stimulants, heavy exercise
- Treat scoliosis
Prognosis
The vital factors in the management of MFS are:
- Diagnosis
- Monitoring of aortic aneurysm and cardiac valves.
- Cardiovascular surgical intervention when the aortic root area/body height ratio becomes abnormal.
- Early diagnosis and surgical management of acute aortic dissection.
- Beta-blocker control and surgery when indicated.
Complications
During Pregnancy – Aortic dissection or rupture may occur when the aortic diameter becomes abnormal. Multidisciplinary approach is recommended.
Pulmonary – Pneumothorax may occur. Lung expansion may be restricted due to pectus excavatum or scoliosis. MFS increases the risk of sleep apnea.
Orthodontic – Maxillary narrowing resulting in high-vaulted palate and mandibular retrognathia resulting in malocclusion is the most common features.



{kind=link}
Comentários
Enviar um comentário