Pediatric Pulmonology: Cystic Fibrosis (CF), Bronchopulmonary Dysplasia (BPD), Diaphragmatic Hernia and more
Table of Contents
Overview
Pediatric patients will commonly present to the ED or clinic with respiratory disease. Most common etiologies are from infection, acute causes, atopy, congenital disease, or genetic disease. This article will focus on Congenital and Genetic causes of respiratory disease in the pediatric population. Congenitally refers to a disease that is present at birth or diagnosed within the first few hours of life. The genetic disease results from alterations within the genome due to mutations or deletions of DNA. Due to the variability of genetic disease, patients will usually present within the first two years of life.
Congenital Disease
Diaphragmatic hernias
Diaphragmatic hernias occur during the fetal developmental period. There are three common presentations called Bochdalek, Morgagni, and Hiatus. Each hernia can result in some pulmonary hypoplasia with changes in circulation and surfactant levels.
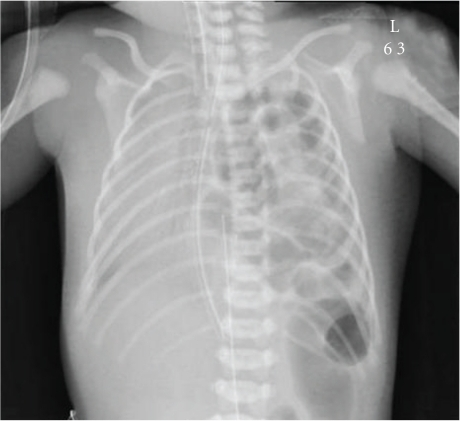
Image: “Chest X-ray shows left-sided congenital diaphragmatic hernia with herniating loops of large bowel into the right hemithorax.” by Arafah M, Boqari DT, Alsaad KO. License: Open Access
Bochdalek hernias are located on the left side of the body. This defect allows for the small and large bowel to enter the thoracic cavity. Left sided hernias are the most common making up 85 % of all pediatric hernia cases.
Morgagni is a less common hernia on the right side of the body. The defect is usually limited and results in liver and large bowel entering the thoracic cavity.
Hiatal hernias result from the stomach sliding into the thoracic cavity through the esophageal hiatus of the diaphragm.
Newborns will present with respiratory distress shortly after birth and can quickly become a medical emergency. Diagnosis is made with chest x-ray and ultrasound to visualize the anatomical changes and to rule out cardiac anomalies and pneumothorax. Treatment for each hernia includes initial management of the airway and then surgery to repair the malformation.
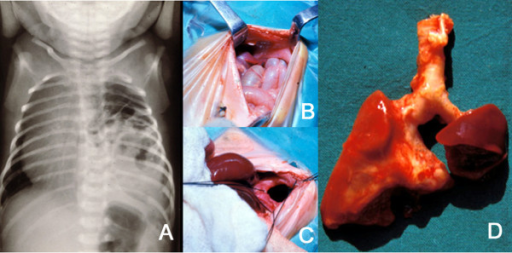
Image: ” A: Plain X-ray of the thorax of a newborn with CDH. There are bowel loops into the left hemi-thorax, the mediastinum is displaced to the contralateral side and the space occupied by the lung is reduced. B and C: At laparotomy, a left, posterolateral diaphragmatic hernia was discovered. In B, small bowel loops can be seen entering the thorax through the orifice. In C, this is seen after reducing the contents of the hernia. D: The patient died of severe persistent pulmonary hypertension days later. At autopsy, extreme left lung hypoplasia and less severe right lung hypoplasia were discovered.” by Tovar JA. License: CC BY 2.0
Congenital cystic adenomatoid malformation (CCAM)
The congenital cystic adenomatoid malformation is a made up of several malformations of the patient’s airway that are associated with adenomatous overgrowth of terminal bronchioles and cystic changes.
With the advent of ultrasound, CCAM has often detected early along in the pregnancy and management can be done in the prenatal stages. Due to the size of some of the cysts, CCAM can lead to hydrops fetalis, a condition where fluid accumulates within fetal compartments.
Postnatally patients may present with respiratory distress. Management depends upon the severity of disease while most cases are benign in severe cases early induction of labor and NICU care are needed until the malformation can be corrected.
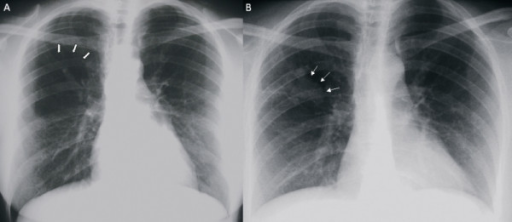
Image: “(a) Posteroanterior chest X-ray showing an area of over-inflation and parenchymal oligemia downstream at the right upper lobe (thick arrows). (b) Posteroanterior chest X-ray showing right parahilar superimposed opacity (thin arrows).” by Discioscio V, Feraco P, Bazzocchi A, Femia R, Romeo C, Fasano L, Pacilli AM, Zompatori M. License: CC BY 2.0
Surfactant deficiency
Surfactant deficiency is a condition that presents in premature babies. Surfactant or Phosphatidylcholine is produced in the lungs by type 2 pneumocytes which allow for the alveoli to stay open and remain compliant by maintaining surface tension. At 32 weeks gestation, fetal lungs are producing enough surfactant to allow for their lungs to stay open.
Respiratory distress syndrome occurs in premature infants when they are born before that 32-week mark. The lack of surfactant results in alveoli that are collapsed and lungs that are not compliant. If premature birth is likely to happen due to medical conditions mothers can be given a dose of steroids which helps to increase the surfactant levels.
The newborns will often be sent to the NICU for airway management and outcomes generally are inversely proportional to age with younger babies being less likely to survive.
Pediatric Pulmonary Hypoplasia
Pediatric Pulmonary Hypoplasia is underdevelopment of the lung that can result in a spectrum of malformations depending upon the developmental time point.
The most common causes are related to volume and pressure within the amniotic sac and lungs. During development, the fetus produces urine from the kidneys which become the amniotic fluid. The fluid is then swallowed and inspired into the lungs which allow for their expansion and aids in their development.
Changes in the amount of fluid can result in lung hypoplasia since the lungs are not expanding. Lung hypoplasia carries a high mortality rate that can be greater than 70 % during the perinatal period. There is often an association with a congenital disease, such as cardiac, skeletal, genitourinary.
Management involves steroids to maximize surfactant and airway support.
Bronchopulmonary dysplasia
Bronchopulmonary dysplasia is a chronic lung condition that results in neonates due to prematurity combined with oxygen and positive pressure ventilation.
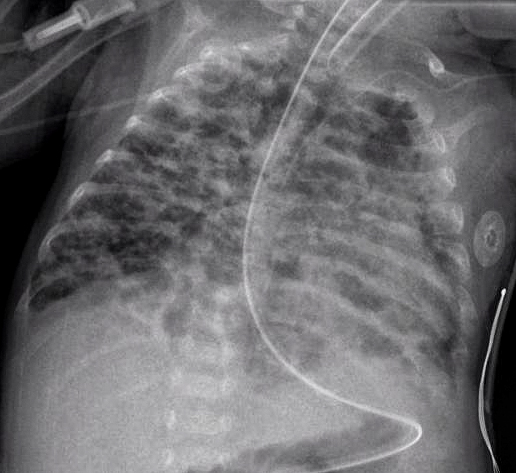
While the exact cause is not known, the leading theory suggests that when babies are born prematurely their lungs are still in an active developing state and when exogenous oxygen is supplied the lungs shift to a mature state leaving the alveolar and vascular premature.
Patients will present in respiratory distress which includes tachypnea, tachycardia, grunts, nasal flaring, intercostal retraction with breaths, and oxygen desaturation.
Imaging studies combined with lung function tests are used to make the diagnosis and grade the severity of the dysplasia. Management includes surfactant replacement, oxygen therapy, and ventilation. Outcomes depend upon the prematurity of the infant.
At 23 weeks there is 25—50 % chance of survival with intervention and these numbers increases with weeks.
Genetic Disease
Cystic Fibrosis (CF)
Cystic Fibrosis is a lethal inherited disease that is most commonly seen in Caucasians. On average 1 in 20 whites are carriers and thus there is the stratification of incidence with higher incidences in northern Europe and very low incidence in Asia. It has been documented for thousands of years in human history with descriptions of salty skinned babies who would have a poor health prognosis.
CF is believed to be a protective mutation against cholera in the heterozygous form. Exocrine glands are affected by a mutation in the CFTR chloride channel. The disruption of Cl- ion flow results in the reabsorption of sodium and water back into the extracellular space. This results in changes to mucus consistency making it thicker and more prone to bacterial growth.
There are multiple alleles of the CFTR gene that influence its penetrance. CF is inherited in an autosomal recessive fashion and both parents will need to be carriers for the disease to present. The most common and severe organs affected by CF are the lungs and the exocrine pancreas. CF’s lethality results from end-stage lung disease.
Since there are multiple alleles of CF presentation times can vary from at birth to later years which result in a diagnosis age of 6 to 8 months.
Signs and symptoms vary by patient and are best divided up by system. Common GI symptoms are meconium ileus, distension of the abdomen, bowel obstruction, failure to thrive, steatorrhea with foul smelling flatulence, jaundice and bowel bleeds.
Common respiratory symptoms are a cough, wheeze, pneumonia, chest pain and exertional dyspnea.
Common genitourinary symptoms include in males undescended testicles, hydroceles, often times they will be infertile due to a failure of the vas deferens to develop. In females, they can present with amenorrhea.
USMLE pearl: The meconium ileus is where the meconium, the first bowel movement from the newborn, is more viscous and results in obstruction of the intestines. It can pass on its own but often times an enema is needed to help it pass. It should be differentiated from Hirschsprung disease.
Diagnosis of CF a based upon genetic testing for mutations in CFTR and with a positive sweat chloride test. One additional finding is required which can include COPD, exocrine pancreas insufficiency, or a positive family history.
In the US babies are all tested for CF after birth to help start with management early as that has been shown to improve outcomes.
On the chest, x-ray patients will show hyperinflation, infiltrates and in some cases bronchiectasis. These are more often seen in the upper lobes.
There is no cure for CF and the management goals are to have a strong doctor-patient relationship in order to maintain lung function with vigilant treatment of infection and clearing of airway mucus; nutritional therapy with supplements of pancreatic enzymes, a supplement of vitamins and minerals.
Maneuvers to tap out mucus are used at home and are often combined with physical therapy.
Medications are bronchodilators, mucolytic agents (dornase alfa), antibiotics such as fluoroquinolones.
Surgical intervention is used for bowel obstructions, gastrostomy tube, and in end-stage lung disease lung transplants can be performed.
The median age of survival globally is 36.9 years with some disparity between men and women. Survival age is higher in America than in other countries around the world. Low socioeconomic status has been shown to reduce lifespan and has been a big factor in how management is done now with frequent follow-ups to ensure compliance.
Alpha 1 antitrypsin
Alpha 1 antitrypsin is an inherited genetic disease due to a loss of the protease inhibitor SERPINA1 located on chromosome 14. It is most prevalent in the white population and has an equal ratio between sexes.
Pediatrics patients will most commonly present with liver pathologies such as jaundice or cirrhosis. This occurs because the mutated form of Alpha 1 antitrypsin is produced in the liver and cannot be secreted into the bloodstream and damages the tissue. A small percentage of pediatric carriers will present with liver pathology due to the different mutant allele.
As the patient’s age, they become more at risk for respiratory pathology. Normally the secreted alpha 1 antitrypsin will circulate to the lungs and break down the neutrophil protease enzyme elastase. Since this does not happen the alveolar walls begin to break down and emphysema results.
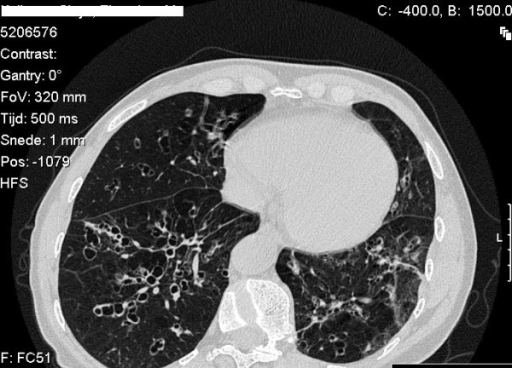
The emphysema is referred to panacinar which is located in the lower lobes of the lung. You can distinguish emphysema from alpha 1 antitrypsin and smoking since smoke rises the damage will be shifted towards the apex of the lung while alpha 1 antitrypsin patients will have it localized towards the lung bases.
Treatment includes trying to optimize lung function by avoiding smoking and regular follow-ups with their doctor.
Emphysema is the most common cause of death followed by liver disease in these patients.
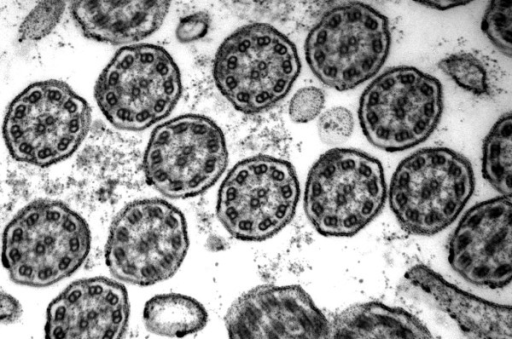
Primary Ciliary Dyskinesia
Primary Ciliary Dyskinesia is often referred to as Kartagener’s syndrome and it is a rare genetic disease that has an autosomal recessive inheritance pattern.
Cilia are proteins that function to help expel mucus and other contaminants from the lung and other tissues. Cilia line the large airways, sinuses, middle ear on the pseudostratified columnar epithelium.
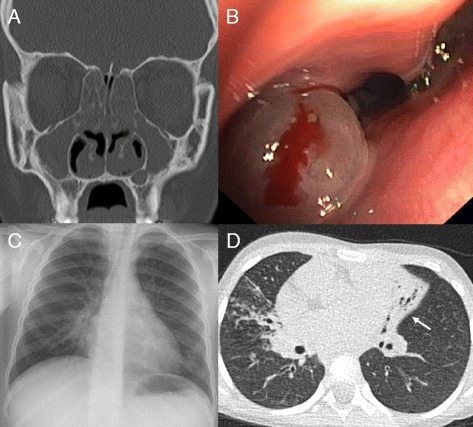
Image: “Clinical features of primary ciliary dyskinesia. (A) Coronal computed tomography (CT) scan of a 17-year-old PCD individual showing diffuse pansinusitis with mucosal thickening and polyposis. (B) Endoscopic view showing nasal polyp (same patient). (C) Chest X-ray of a 6-year-old PCD individual with middle lobe atelectasis. Silhouetting of the right heart border is present. (D) Chest CT of a 6-year-old individual with situs inversus totalis. The left-sided middle lobe shows extensive bronchiectasis with volume loss (white arrow). In addition, consolidations and mucous impaction are present in the right upper lobe.” by Werner C, Onnebrink JG, Omran H. License: CC BY 4.0
Due to the abnormal ciliary function patients will present with an increased number of infections of the respiratory tract, sinuses, and otitis media. In addition, male infertility occurs due to motility problems of the sperm since the male sperm cell’s tail has a similar core structure to cilia and are affected by the mutation.
There is also an association with situs inverses a condition where the internal organs are flipped within the body (for example the heart faces to the right instead of the left which is also referred to as dextrocardia).
Management includes optimizing lung function by avoiding smoking and following up with their physician on a regular basis. Due to the increased number of infections keeping up to date with vaccinations is important particularly for Haemophilus influenzae Type B, Flu, and MMR.



{kind=link}
Comentários
Enviar um comentário