Spinal Muscular Atrophy (SMA) in Children — Types and Treatment
Table of Contents
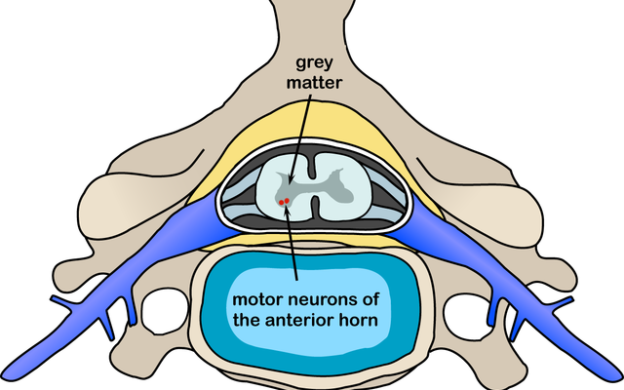
Overview
Spinal Muscular Atrophy presents early in infancy with hypotonia and weakness and is considered the most common monogenic cause of infant mortality. This condition leads to the breakdown of the nerve cells in the lower part of the brain, as well as the spinal cord. Due to the death of the cells, the brain is unable to send signals to the body muscles of the infant.
Infants suffering from this condition experience challenges and difficulties in crawling, walking, as well as breathing. For older kids, they face a challenge in getting around the everyday activities, such as combing their hair or brushing their teeth.
Several studies have been conducted to identify the genes responsible that cause SMA; however, at the moment, there is still no cure for this condition. Despite the several limitations, hundreds of kids have learnt to live independently through physical as well as occupational therapy.
Clinical picture and phenotypes of SMA
The disease can present prenatally, such as type 0, or during the infantile period type 1 or later in life as in type 2 or 3. Type 4 presents in adults with less severe manifestations. Type 0 and type 1 are the most severe forms. SMA presents with symmetrical weakness and hypotonia in all extremities, but more in the lower limbs. Deep tendon reflexes are absent. Respiratory muscles can be affected with more severe forms leading to restrictive respiratory insufficiency. Associated manifestations include congenital heart anomalies and sleep abnormalities.
SMA 0
SMA 0 is the most severe form with the least number of SMN gene copies, usually 1. It presents prenatally with a decrease in fetal movements and progressive hypotonia postnatally. Infants are born with multiple skeletal deformities as a result of hypokinesia in utero e.g. joint contractures, polyhydramnios, and pulmonary hypoplasia. Death usually occurs before 6 months from respiratory failure.
SMA 1 (Infantile)
Also called Werding-Hoffmann, SMA1 is the disease with 2—3 SMN gene copies. It presents early in infancy with progressive symmetrical flaccidity in all extremities manifests as frog leg deformity. Bulbar muscles weakness results in a weak cry, poor sucking, and drooling of secretions from the mouth. Paradoxical breathing and a bell-shaped chest are the results of the weakness of intercostal muscles. Death usually occurs before 2 years of age from respiratory failure. Cardiac anomalies are not common in SMA type 1. This condition begins to affect the infants from birth up to 6 months of age, with most babies showing signs of the disease by 3 moths. Some rare cases of the manifestation of the disease before birth have been reported.
SMA 2 (Intermediate)
SMA 2 is also called Dubowitz disease. It presents between 6—18 months in a less severe form than type 0 and 1. Infants present with delayed sitting, but without standing unsupported. Later on, they become unable to sit independently. Limb weakness is also evident with tremors. Other manifestations include; absent deep tendon reflexes, respiratory muscle weakness, and tongue atrophy. Patients usually die within 25 years due to restrictive lung disease from respiratory muscle weakness and progressive scoliosis. The progress of this type is known to vary from one individual to the next; some people gradually grow weaker with time, while others can contain any progress of the disease. The life expectancy of infants is reduced; however, most of them live well into adulthood despite having weak muscles and respiratory problems.
SMA 3 (Juvenile)
SMA 3, also called Kugelberg-Welander disease, presents later in life, mostly after the 12thmonth of age, Patients that can stand and walk independently lose it later. Falls are common with foot deformities and muscle weakness. A normal life expectancy is common, but patients become wheelchair dependent. Respiratory involvement is rarely noticeable, and the life expectancy is normal or nearly normal.
SAM 4 (Adult-onset)
Patients have 4—8 copies of the SMN2 gene. It is the least severe form of SMA and presents late in life. Patients have a normal life expectancy and normal motor development.
Differential Diagnoses of SMA
Differentiation of SMA type 0 and 1
SMA type 0 and 1 should be differentiated from early causes of infantile weakness or paralysis, including congenital myasthenia gravis, congenital myotonic dystrophy, traumatic myelopathy, myopathies, X-linked infantile spinal muscular atrophy and spinal muscular atrophy with respiratory distress type 1.
X-linked infantile SMA
X-linked infantile spinal muscular atrophy is due to X-linked mutation of the ubiquitin activating enzyme 1 leading to hypotonia, absent reflexes, and fractures.
Spinal muscular atrophy with respiratory distress type 1
Spinal muscular atrophy with respiratory distress type 1 is due to the mutation in the immunoglobulin binding protein 2 gene. It is characterized by respiratory distress due to diaphragmatic paralysis in infants less than 6 months. Infants are ventilator dependent early in life.
Metabolic disorders
Metabolic disorders are associated with hypotonia and weakness include
Glycogen storage diseases, characterized by hypotonia but also hypertrophic cardiomyopathy, macroglossia, and hepatomegaly.
Prader-Willi syndrome
The Prader-Willi syndrome is characterized by hypotonia and bulbar muscle weakness, as well as genital hypoplasia. The hypotonia improves with age.
Differentiation of SMA type 2 and 3
Type 2 and 3 SMA should be differentiated from other causes of hypotonia in late infancy and early childhood. Muscular dystrophies, Guillain-Barré syndrome, and myasthenia gravis.
Muscular dystrophies
Muscular dystrophies including Duchenne and Becker are associated with proximal muscle weakness with cardiomyopathy and dysrhythmias. Patients present with calf pseudohypertrophy, a waddling gait and lumbar lordosis.
Limb-girdle muscular dystrophy
Limb-girdle muscular dystrophy affects mainly girdle muscles of the shoulder or pelvic without facial or extra ocular muscle affection. The distal muscles are usually spared with minimal or no weakness.
Differentiation of SMA type 4
SMA type 4 should be differentiated from causes of hypotonia in adults, including amyotrophic lateral sclerosis and spinobulbar muscular atrophy. Amyotrophic lateral sclerosis is characterized by both upper and lower motor neuron manifestations of weakness, and atrophy,
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis is characterized by both upper and lower motor neuron manifestations of weakness, atrophy, fasciculations, spasticity, and hyperreflexia.
Bulbar muscle weakness
Bulbar muscle weakness is also evident. Spinobulbar muscular atrophy presents with weakness, atrophy of limbs and facial muscles as a result of X-linked disease. It is also characterized by endocrinal dysfunction in the form of androgen resistance and gynecomastia.
Diagnosis of SMA
Electromyography is used to detect spontaneous twitches and sharp waves. Polyphasic action potentials with high amplitude are characteristics. Muscle biopsy shows atrophic muscle fibers with hypertrophied fascicles that are innervated by the remaining nerve fibers. Sequencing of the SMN 1 gene to detect homozygous deletions or point mutations of SMN gene is confirmatory.
Diagnosis tests
- Pre-implantation testing: Used to screen for SMA-affected embryos during in-vitro fertilization.
- Prenatal testing: Screens for SMA through chorionic villus sampling, as well as cell-free fetal DNA analysis.
- Carrier testing: The test uses saliva or a blood sample to make an analysis of whether the offspring’s of SMN1 carriers will be affected by the disease.
Management of SMA

Image: “Max High named Firefighter for the Day” by North Charleston. License: CC BY-SA 2.0
Supportive therapy is the mainstay of treatment for patients with spinal motor atrophy. Respiratory support, prevention of complications, follow-up with the nutritional needs and physical therapy are mandatory for all patients. Respiratory muscles weakness and deformities lead to restrictive respiratory failure and retention of secretions. Postural drainage of the secretions with chest physiotherapy helps with clearance of the secretions from the chest.
Respiratory support with nasal ventilation and sometimes mechanical ventilation are utilized in children with respiratory insufficiency. Scoliosis can be prevented with spinal bracing, while physical therapy should be used early to prevent muscular deformities and joint contractures.
Dysphagia, drooling of saliva, poor feeding and risk of aspiration are all manifestations of bulbar dysfunction in SMA patients necessitating the need for nutritional support. Nutritional support is mandatory for SMA type 1 and 2 since they commonly develop bulbar muscular dysfunction. Nutritional support includes prevention of aspiration, a liquid diet or even gastrostomy tube feeding.
Mobility support using an orthotic device should be used to support the body and to facilitate the walking of the patient.
Nusinersen
Nusinersen is a novel drug that was approved most recently, and it is designed to increase the expression of the SMN protein leading to less progression of symptoms. The drug was noted to improve the motor development in children with delayed milestones in spinal motor atrophy compared to control patients. The drug can be injected intrathecally in both children and adults with SMA. Adverse effects include constipation, coagulopathy, thrombocytopenia, nephrotoxicity and respiratory infection.



{kind=link}
Comentários
Enviar um comentário