Neuromuscular Disease: Myasthenia Gravis, Guillain-Barré Syndrome, Duchenne Muscular Dystrophy, ALS and More
Table of Contents
- Overview
- Classification of Neuromuscular Diseases
- Amyotrophic Lateral Sclerosis (ALS)
- Management of Neuromuscular Diseases
- Duchenne Muscular Dystrophy (DMD)
- Becker Muscular Dystrophy (BMD)
- Guillain-Barré Syndrome (GBS)
- Myasthenia Gravis (MG)
- Lambert-Eaton Myasthenic Syndrome (LEMS)
- Spinal Muscular Atrophy (SMA)
- Myotonic Dystrophy (DM)
- Charcot-Marie-Tooth (CMT) Disease
- Spinal and Bulbar Muscular Atrophy
- References
Image: “Campylobacter jejuni, which triggers about 30% of cases of Guillain-Barré syndrome”. License: Public Domain
Overview
Neuromuscular disorders usually lead to impaired movement, muscle weakness, fatigue, twitches and rigidity. This group of disorders can be genetic or acquired. Upper motor neuronal dysfunction lead to spasticity and hyperreflexia while lower motor neuronal lesions lead to paralysis with muscle fasciculations.
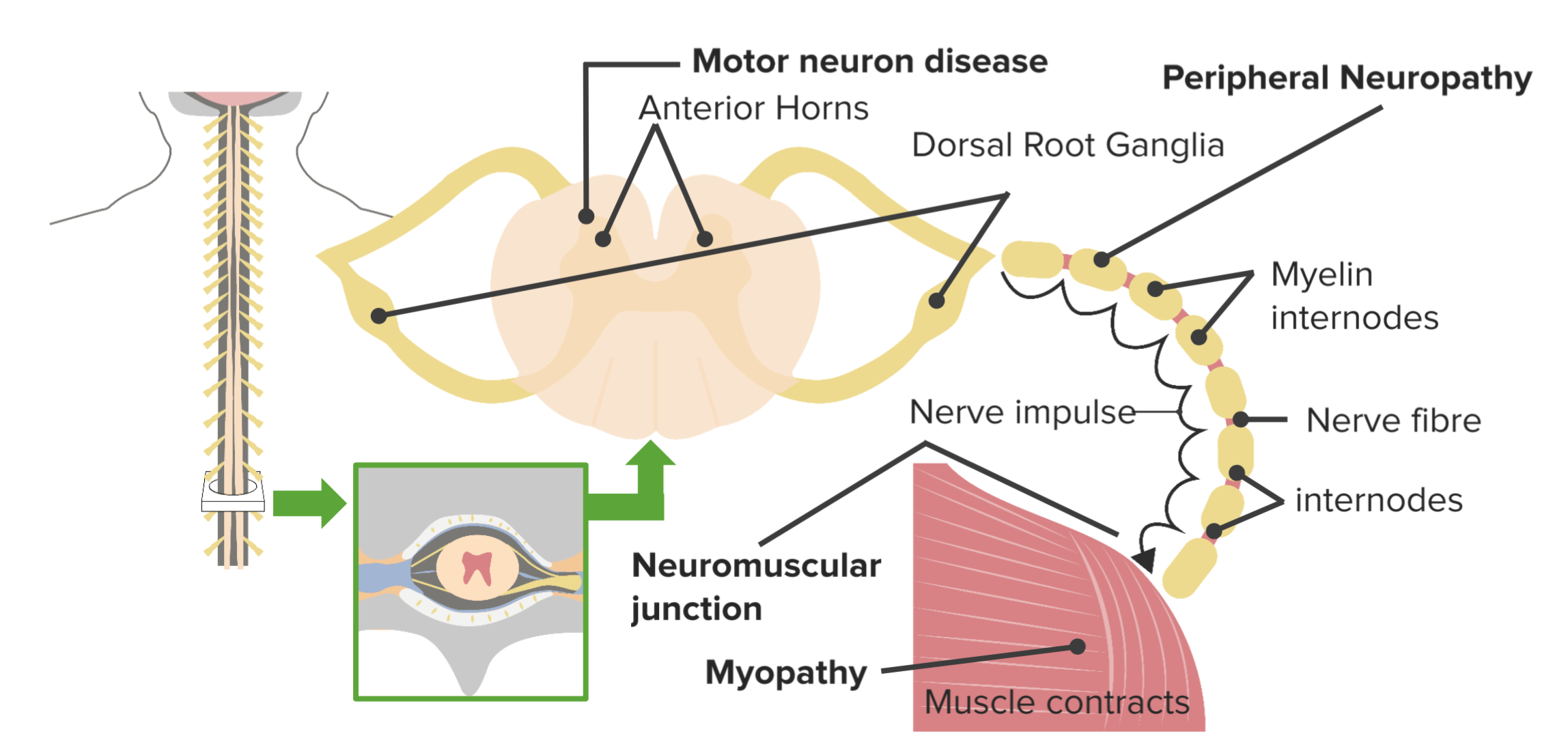
Classification of Neuromuscular Diseases
- Upper motor neuronal disorders that lead to hypertonia and hyperreflexia. Some upper motor neuron diseases affect the brain and include stroke, Parkinson’s disease, Huntington’s chorea, Creutzfeldt-Jakob disease and multiple sclerosis (MS).
- Lower motor neuronal lesions that lead to paralysis with muscle fasciculations.
Amyotrophic lateral sclerosis is an example of both upper and lower motor neuron lesions.
Muscular disorders include a group of genetic muscular dystrophies e.g. Duchenne’s andBecker’s muscular dystrophies and mitochondrial dystrophies.
Neuromuscular junction disorders involve a group of disorders e.g. myasthenia gravis,Lambert-Eaton myasthenic syndrome, tetanus, and botulism.
Peripheral nerves can be involved with neuropathy, trauma, and demyelinating diseases e.g. Charcot-Marie-Tooth (CMT) disease. The following are a few examples of neuromuscular disorders that affect the muscles, nerves, or upper motor neurons.
Amyotrophic Lateral Sclerosis (ALS)
Definition
ALS is a fatal degenerative disorder that is characterized by muscle weakness and disabilitydue to both upper and lower motor neuron lesions.
Epidemiology
The disease is usually sporadic but is less commonly genetically inherited. It is the most common disorder of the motor neuron system. It is a fatal disease with a median survival rate of 3 years, but this can be extended with the right treatment.
Pathology
The upper motor lesions are due to involvement of neurons in the corticospinal tract of the spinal cord, medulla, pons, internal capsule and Brodman area 4 in the cortex while the lower motor lesions are due to involvement of the motor nuclei in the brain stem and spinal cord.
Clinical presentation of ALS
Initial presentation of ALS is asymmetric extremity weakness that affects the upper or lower limbs. Foot drop or lateral hand weakness and atrophy are the initial common presentations.
Weakness progresses to involve axial trunk muscles with head drop or bulbar muscles with dysarthria and dysphagia, respiratory muscles are affected in rare cases. Upper motor neuron manifestations include spasticity, hyperreflexia and ankle clonus. Bulbar upper motor neuron symptoms include dysphagia and dysarthria. Sometimes, pesudopulbar affect, which is inappropriate laughing or crying spontaneously triggered by stimuli, occurs. Lower motor neuron manifestations include weakness, muscular atrophy and fasciculation.
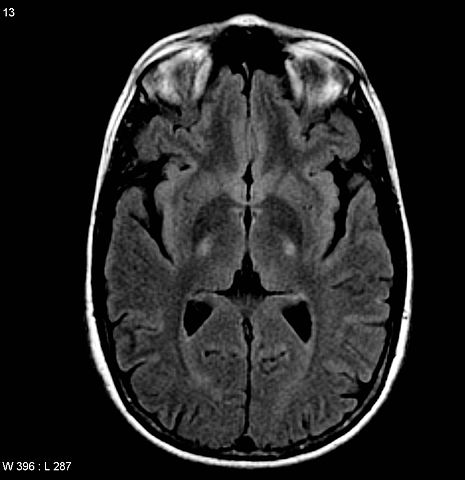
Image: “Amyotrophic lateral sclerosis. MRI (axial FLAIR) demonstrates increased T2 signal within the posterior part of the internal capsule, consistent with the clinical diagnosis of ALS” by Frank Gaillard. License: CC BY-SA 3.0
Autonomic manifestations present with urinary urgency, constipation and sweating. Cognitive impairment with dementia can also be found in up to 50% of patients.
Diagnosis is clinical with careful history and clinical examination. Electromyography can be applied with muscle biopsy and neuroimaging to exclude other causes of upper or motor neuron disease. Electromyography results will show combined acute and chronic denervation.
Management of Neuromuscular Diseases
It involves management of acute occurrences with respiratory support, pain medications and muscle relaxants.
- Long term therapy involves pain control, muscle relaxation and physiotherapy.
- Riluzole is proven to delay the progression of the disease and hence the need for ventilation support and thus improves survival for few months.
- Enteral nutrition via cutaneous gastrostomy tube is encouraged to boost patients weight.
- Creatine and high dose vitamin E should be avoided.
Management with respiratory support, physical therapy, pain medications and muscle relaxants can help as palliative therapy for patients with ALS. Riluzole is proven to delay the need for ventilation support and improve survival for few months.
Duchenne Muscular Dystrophy (DMD)
This is an X-linked recessive muscular dystrophy due to dystrophin gene mutation.
Epidemiology
The disease manifests early in the first 2 years of life with motor developmental delay and pelvic girdle weakness. The symptoms worsen quickly over time.
Clinical Presentation
Early manifestations include delayed walking, difficulty rising from a seated position or climbing up the stairs. This is because the disease affects the lower limbs earlier than the upper limbs and proximal muscles earlier than distal muscles. Repeated falls and bone fractures are common among almost all patients with DMD. Cognitive impairment with behavioral dysfunction is also common.
Cardiac involvement: the disease progresses to dilated cardiomyopathy, mitral regurgitation and different types of arrhythmias.
Respiratory failure due to muscular weakness and scoliosis are primary causes of death in DMD patients.
Signs of DMD
Pseudohypertrophy of the calf muscles is noted in most patients while other voluntary muscles show weakness and wasting. Lumbar lordosis, waddling gait, and Gower’s sign where the patients use their hands for support while standing up are also common.
Serum creatine kinase is markedly elevated than normal range and muscle biopsy with genetic testing are confirmatory for the diagnosis. Muscle biopsy shows degeneration and regeneration with fibrosis, fat, and connective tissue deposition in the muscles.
Becker Muscular Dystrophy (BMD)
This is an X-linked recessive muscular dystrophy that affects the dystrophin gene. The disease mainly affects male patients and is like DMD but can be distinguished from the disease because Becker dystrophy presents later in life with milder but variable severity.
Patients have preserved motor power and activity until later in life with no or less cognitive impairment compared to DMD.
Cardiomyopathy is more evident in patients with BMD, which can be explained by preserved muscular power and activity leading to more work load on the cardiac muscles with abnormal dystrophin.
The diagnosis of the disease is like DMD and depends on :
- Moderate to severe elevation of creatinine kinase.
- Dystrophin gene deletion in 98% of the cases.
- Muscle biopsy with dystrophin antibody staining shows presence of dystrophin in variable percentages.
The disease has no cure and al avenues for treatment involve improvement of the quality of life
Guillain-Barré Syndrome (GBS)
Definition
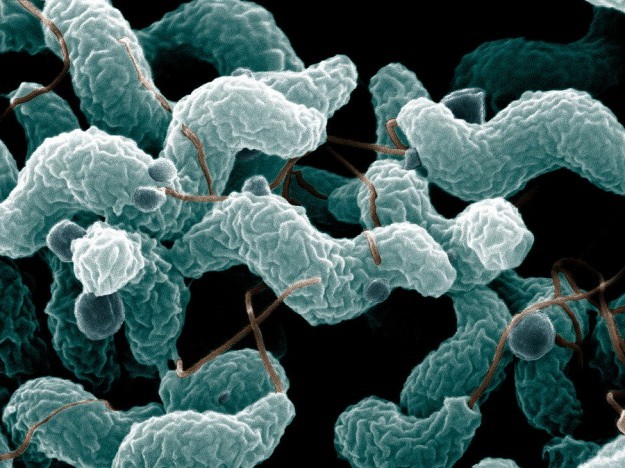
Image: “Campylobacter jejuni, which triggers about 30% of cases of Guillain-Barré syndrome”. License: Public Domain
GBS is an autoimmune disorder with antibodies against peripheral nerves secondary to a preceding infection. An infection with campylobacter jejuni, cytomegalovirus or Epstein-Barr virus is responsible for the formation of antibodies which lead to demyelination of the peripheral nerves.
Epidemiology
The disease affects 1-2 per 100,000 persons in the population with a female to male ratio of 1.5:1. It can affect all ages, but it tends to have a bimodal distribution curve with peaks at 15-35 years and 50-75 years.
Clinical picture of GBS
The disease may be preceded by an acute diarrheal illness or upper respiratory tract infection that triggers the autoimmune process.
After 2-4 weeks, the disorder will manifest as progressive symmetric muscle weakness or paralysis that involves limbs, respiratory muscles, face and bulbar muscles accompanied by diminished or absent reflexes. Respiratory or oropharyngeal involvement requires prolonged ventilatory support and airway protection. Facial nerve paresis, pain and autonomic dysfunction may be present.
Diagnosis
CSF analysis will show albuminocytologic dissociation, which is elevated protein with normal white cell count.
Treatment
Plasmapheresis and immunoglobulin therapy (IVIG) are the main modes of treatment. They have been shown to be of equal efficacy and thus preference is based on availability and clinical status of the patient.
In case of respiratory muscle involvement intensive care therapy and breathing support is recommended.
Physical therapy is indicated in the recovery phase for return of limb function.
Myasthenia Gravis (MG)
MG is an autoimmune disorder with T-cell dependent antibodies against post-synaptic acetylcholine receptors at the motor endplate. The disorder can affect ocular, bulbar, extremity and even respiratory muscles causing weakness and fatigue that fluctuates throughout the day.
Weakness is more evident later during the day or after exercise. Ocular disease presents with diplopia or ptosis while pharyngeal muscle weakness presents with dysarthria and dysphagia. Muscles of the jaw, face, neck, trunk and respiratory muscles are affected in the generalized form of the disease.
Respiratory muscle weakness is known as myasthenic crisis due to respiratory failure which can be complicated by dysphagia and aspiration. Infection, surgery, or immunosuppressant withdrawal can precipitate a myasthenic crisis.
Management of MG
Diagnosis is based on clinical presentation, laboratory tests and imaging. Anti-acetylcholine receptor antibody, anti-striated muscle antibody, anti-muscle specific kinase antibody are all highly sensitive and specific tests for ocular and generalized MG.
Up to 15% of patients with MG will have enlarged thymoma on chest radiographs. Repetitive nerve stimulation and single fiber electromyography will show impaired neuromuscular transmission at the motor endplate. Improvement of symptoms after acetylcholine esterase inhibitors (edrophonium) injection was used as an initial screening test but lacks specificity.
Treatment of MG
Anti-acetylcholine esterase medications are used to control symptoms while immunosuppressive therapy e.g. steroid, azathioprine is used for disease control.
Plasmapharesis and intravenous immunoglobulins are used in severe cases. Some medications can worsen MG weakness including fluoroquinolone, quinine, magnesium, aminoglycoside antibiotics, procainamide and beta blockers.
Lambert-Eaton Myasthenic Syndrome (LEMS)
Definition
This is an autoimmune disease affecting the neuromuscular junction that presents with weakness in the setting of small cell lung cancer. The weakness is due to decreased release of acetylcholine from nerve terminals with normal vesicles in the presynaptic terminals and normal receptors in the postsynaptic terminals. It is thought to be due to antibodies against presynaptic voltage gated calcium channels that allow calcium influx and release of acetylcholine.
Clinical presentation
Patients present with progressive proximal muscle weakness and fatigue. Respiratory failureis common. Ptosis, extraocular muscle paresis and cranial nerve involvement is also common.
Treatment
Administration of guanidine increases presynaptic acetylcholine release resulting in improvement of muscular weakness. Other therapies include pyridostigmine which is an acetylcholine esterase inhibitor, IVIG, azathioprine, which is an immunosuppressant and aminopyridine, which prolongs membrane depolarization.
Spinal Muscular Atrophy (SMA)
SMA is defined as a degeneration of the anterior horn cells of the spinal cord that can manifest prenatally or early in life. There are four types of the disorder; the most severe form is calledWarding-Hoffman disease.
Infants with this disorder usually die from respiratory failure within the first year of life. Patients present with lower motor neuron symmetrical weakness or even paralysis of the proximal muscles with absence of deep tendon reflexes. Bulbar muscle involvement leads to a weak cry, poor suckling, aspiration and tongue twitches.
Respiratory muscle weakness affects the intercostal muscles but not the diaphragm, leading to respiratory failure and paradoxical breathing with the chest wall moving inwards during inspiration.
The diagnosis of SMA is made by electromyography, nerve conduction studies and muscle biopsy. Electromyography shows fibrillation in abnormal spontaneous activity in the muscles while a muscle biopsy shows atrophic and hypertrophied muscle fibers.
Management is mainly supportive for respiratory failure. Nusinersen is a drug approved for treatment of SMA that increases survival of motor neuron protein in affected patients.
Myotonic Dystrophy (DM)
This is an autosomal dominant disorder caused by expansion of the CTG (DM1) or CCTG (DM2) trinucleotide repeat in the dystrophica myotonica protein kinase gene that results in arrhythmia, balding, infertility, cataract and diabetes mellitus.
Cognitive impairment and mental retardation can develop in patients with DM1. Cancer susceptibility is higher in patients with DM specially brain, ovarian, colon and endometrial tumors.
Diagnosis is made using electromyography, muscle biopsy and genetic testing. Treatment is mainly supportive and involves use of pacemakers, respiratory support, physical therapy, CNS stimulants and analgesics depending on the clinical presentation.
Charcot-Marie-Tooth (CMT) Disease
It is also called hereditary motor sensory neuropathy and is caused by genetic mutation of one of the myelin genes. The disease can be classified into type 1 and 2 depending on the mutation. It is a combination of motor and sensory manifestations and deformities due to nerve demyelination.
Patients develop muscle weakness with diminished or absent reflexes that lead to atrophy of the calf muscles and intrinsic muscles of the hands and feet. Deformities develop in the form of Pes cavus foot deformity, stork leg deformity and even kyphoscoliosis. Sensation is also affected causing absent deep proprioception and vibration sensation.
Diagnosis is made through nerve conduction studies, which show slow conduction of 60% normal in both sensory and motor nerves in type 1 CMT while a nerve biopsy shows defective myelination with onion bulbs of demyelination and remyelination with abundant Schwann cells. Genetic testing detects the type of mutation and confirms the diagnosis.
Type 2 CMT has similar presentation of distal motor weakness, sensory loss and diminished reflexes and deformity but is less severe than type 1 and presents later in life.
Management of CMT disease is mainly supportive. Physical therapy and even surgery are indicated for deformities. Ascorbic acid, neutrophin-3 and progesterone antagonists help promote myelination and slow the disease progression.
Spinal and Bulbar Muscular Atrophy
Definition
This group of genetic neurodegenerative disorders that present with muscle weakness and atrophy due to degeneration of motor neurons in the brain stem and spinal cord.
Etiology
Results from genetic mutation in the androgen receptor gene that is inherited in an x-linked recessive manner.
Presentation
This group of genetic disorders presents with muscle weakness and atrophy. Upper and lower motor neuron lesions can both be present due to degeneration of neurons in the brain stemand spinal cord.
Endocrine manifestations are present due to X-linked recessive mutation in the androgen receptor (AR) gene, which leads to androgen insensitivity. Early neuromuscular signs include weakness of the tongue, fasciculations, weakness of the limbs. Later respiratory muscle weakness and action tremors later set in. Endocrine features of gynecomastia, erectile dysfunction, testicular atrophy and subfertility may be evident.
Treatment
There is no known cure and treatment is focused on rehabilitation and slowing of muscle weakness.



Comentários
Enviar um comentário