Polyneuropathies — Duchenne Muscular Dystrophy, Limb Girdle Muscular Dystrophies, Channelopathy
Table of Contents
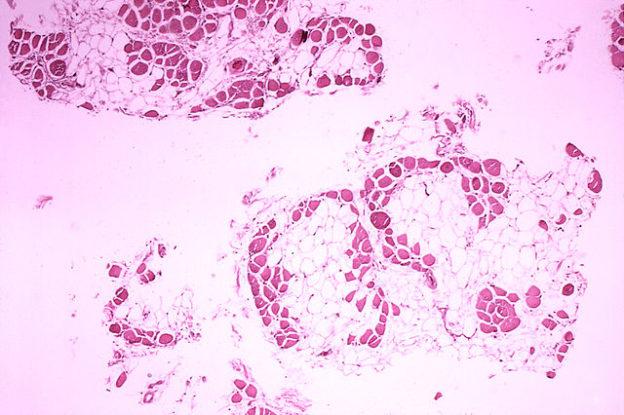
Image : “Duchenne-muscular-dystrophy” by
Dr. Edwin P. Ewing, Jr.. License: Public Domain
Definition of Polyneuropathies
Polyneuropathies are defined as disorders of peripheral and cranial nerves that are widespread and often symmetrical. They often present with both weakness and sensory loss in a “glove and stocking” fashion.
Myopathy is a non-specific term used to refer to a disease of the skeletal muscle which is not related to the disorders of the nerves and neuromuscular junctions.
Dystrophies are a group of non-inflammatory hereditary conditions which show dystrophic changes (increased connective tissue, fiber splitting) on biopsy without degeneration in morphology of muscles. They are not necessarily due to defects of the dystrophin gene.
Myositis implies an inflammatory process.
Image: “Myopathy junction” by lecturio
Causes of Polyneuropathies
- Immune-mediated
- Metabolic
- Hereditary
- Toxic
- Infectious
- Endocrine
- Drug induced
Dystrophinopathies
Dystrophinopathies include a range of muscular disorders due to pathogenic variations in the gene coding for the protein dystrophin. Dystrophin is provides strength to the muscle fibers and without dystrophin muscles are prone to injuries and destruction.
Duchenne muscular dystrophy
This is an X-linked recessive condition that is caused by a deficiency of the protein dystrophin. Dystrophin is involved in the maintenance of the cellular cytoskeleton. DMD results from a mutation in this gene resulting in a non-functional protein. Patients usually present between the ages of 3 and 5. The image below shows the dystrophin complex.
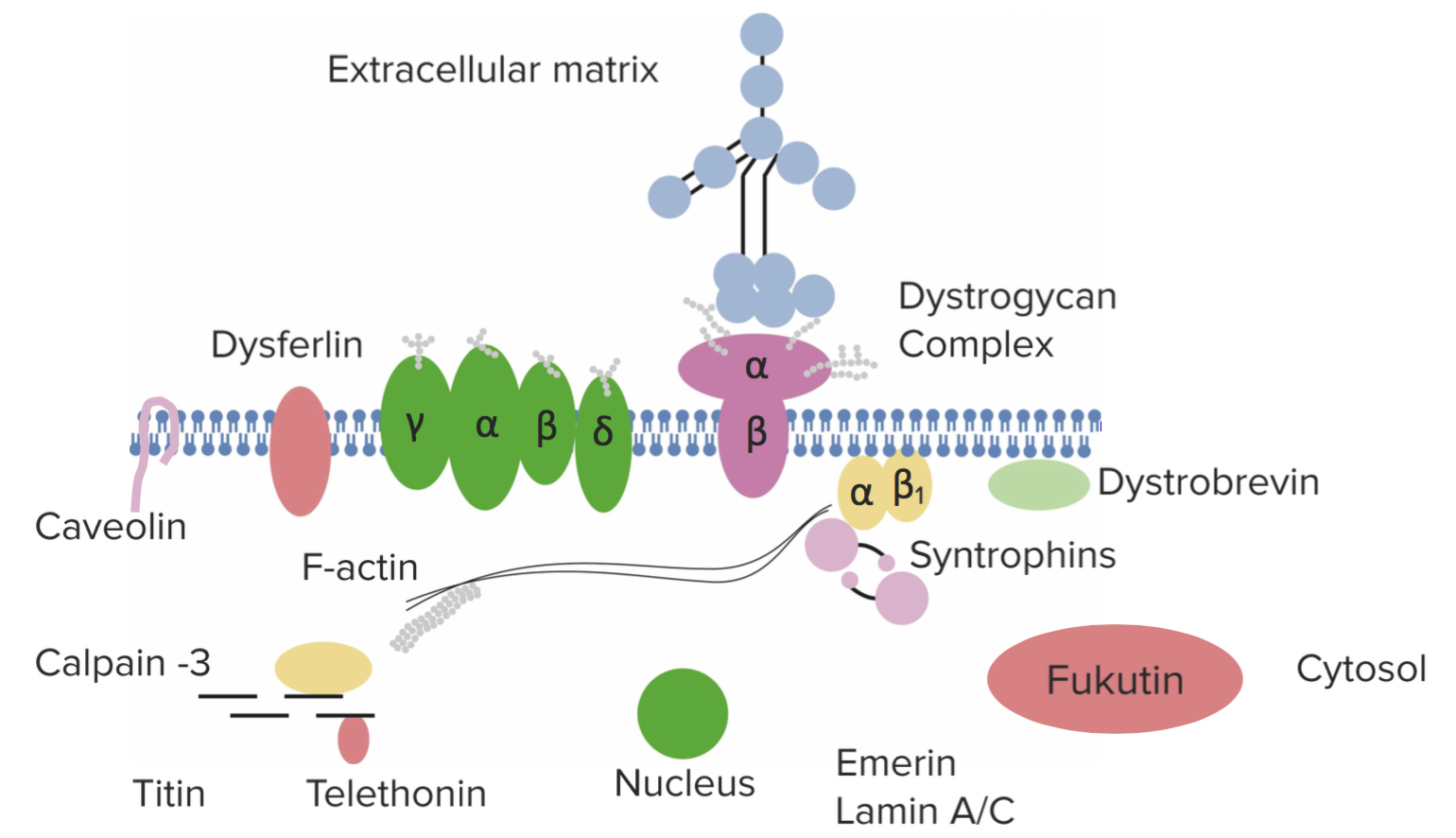
Diagnosis
As a pediatric condition, parents will often come complaining that their child is clumsy or grows excessively tired easily.
These patients will have difficulty walking (and will find it even harder to walk on their toes). They are usually unable to stand without using their own body to steady themselves and, as such, present with Gower’s sign (where patients use their own hands to push off the thighs when rising from the floor). They may also waddle.
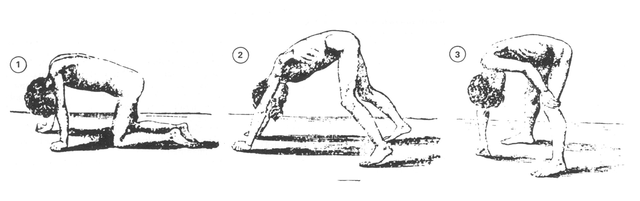
Image: “Gower’s sign.” by William Richard Gowers (1845–1915) – Gowers WR. Clinical lecture on pseudohypertrophic muscular paralysis. Lancet 1879;ii,73-5. License: Public Domain
On examination, you may find pseudohypertrophy of gastrocnemius (calf) muscles. Examine the mental state of the child as retardation is common.
A definitive diagnosis is usually made by a biopsy and negative dystrophin immunostain. Creatinine kinase will be raised (from muscle degeneration). EMG will show “polyphasic” potentials. The biopsy can also show necrotic muscle fibers.
Treatment
Physical therapy can maintain self-ambulation, although this is an inevitability. Patients can survive around 20 years, but the condition is fatal in 100% of cases.
Becker muscular dystrophy
This has an incidence of 0.3/1,000 live male births. This is an X-linked recessive condition and is also caused by a mutation in the dystrophin gene. In this case, the dystrophin protein has some limited function. It presents at a later age than Duchenne, and is associated with a better prognosis.
Diagnosis
These patients will present later than those with Duchenne muscular dystrophy. They are usually between 5 and 15. On Western blot, their dystrophin levels will appear normal (although the protein itself is abnormal) – this is in comparison to DMD, which has a significantly decreased dystrophin on Western blot.
The symptoms are similar to DMD but often milder, and patients are less likely to have mental retardation and have a better prognosis (30-40 years instead of teens).
Limb-girdle Muscular Dystrophies
These are a heterogenous group of inherited dystrophies typically inherited in an autosomal dominant/recessive pattern. Mutations can occur in a number of muscle-related proteins.
Diagnosis
When patients present with muscular weakness in their shoulders and pelvic girdle between the ages of 10 and 20, suspect a Limb-girdle muscular dystrophy.
These autosomal dominant conditions are less severe than Beckers or DMD, but are severely disabling within 25 years of onset. Patients usually will not have hypertrophy of the muscles involved and the symptoms spare ocular and bulbar muscles. Cardiomyopathy is rarely seen.
On investigation, expect to find an elevated creatinine kinase. Biopsy and genetic screening can provide a definitive diagnosis.
Myotonic Dystrophy
Myotonic dystrophy belongs to a class of disease known as the Trinucleotide repeat expansion diseases, which includes Huntington’s disease, Friedreich ataxia, and Fragile X syndrome. Myotonic dystrophy results from a trinucleotide expansion of CTG. It is found on chromosome 19 and is inherited in an autosomal dominant pattern. It affects the Cl-channel and is thus a channelopathy.
Diagnosis
This is the most common form of adult-onset muscular dystrophy. It presents in two different types. Type 1 is known as Steinert’s disease, whilst type 2 is known as proximal myotonic myopathy (PROMM).
In myotonic dystrophy, there is marked distal weakness. Patients will have myotonic (a state of increased muscle contraction and impaired relaxation). Some develop frontal balding, cataracts and cardiac conduction defects as this is a multi-system disease.
Investigations will find a normal or mildly elevated creatinine kinase (a marker of muscle degeneration). DNA testing can be done to make a definitive diagnosis. EMG can also be performed, where myotonic discharges will be seen.
Complications of Muscular Dystrophies
Disability and dependence on walking aids like wheelchairs due to muscular injuries and contractures. Osteopenia and osteoporosis due decreased mobilization and immobilization. This can give rise to fractures and scoliosis. Cardiac failure due to cardiomyopathies.
Prognosis of Muscular Dystrophy
Though there has been much advancement in the treatment of muscular dystrophy but the disease is still incurable and non-preventable. Most of the patients die by the age of 30 years due to cardiopulmonary failure. Patients can have a better quality of life with proper care.
Mitochondrial Myopathies
These are a clinically and genetically heterogenous group of disorders that include mitochondrial encephalopathy with lactic acidosis and stroke-like symptoms (MELAS), mitochondrial encephalopathy ragged red fibers (MERRF), progressive external ophthalmoplegia (PEO) and Kearns-Sayre syndrome.
These are caused by mutations on the mitochondrial DNA and, as such, are either maternally inherited, or caused by sporadic mutation of the mitochondrial DNA.
Diagnosis
These disorders are commonly associated with neurological manifestations like seizures, strokes and migraine. A muscle biopsy will show ragged-red fibers which contain accumulations of glycogen and neutral lipids.
Inflammatory Myopathies
These are a group of diseases including polymyositis, dermatomyositis, inclusion body myositis and necrotizing autoimmune myopathy. There is no known cause for inflammatory myopathies. These result in chronic inflammation and weakness of muscles.
Inclusion body myositis (IBM) is caused by the accumulation of tau aggregates not in the central nervous system. It is often termed a peripheral tauopathy.
Polymyositis is an autoimmune-mediated striated muscle inflammation.
Dermatomyositis has a similar presentation to polymyositis, except with involvement of the skin. It affects patients between 50 and 70, with a higher incidence in both females (2:1 female:male) and African Americans.
Inclusion body myositis
Patients are typically older than 50. On examination, patients often have weakness in their quads, fingers or pharyngeal muscles to begin with. Ventral muscles of the extremities are more affected than dorsal muscle groups. Around 3% of patients become wheelchair dependent.

Image: “Polymyositis: Inflammatory infiltrates in a muscle biopsy. Holes in the muscle cell vacuoles, deposits of abnormal proteins within the cells and in filamentous inclusions (hence the name inclusion body myositis} are clearly visible in the cellular interstitial space.” by Jensflorian – Own work. License: CC BY-SA 3.0
On histological investigation, ringed vacuoles and intranuclear inclusion are seen. There is no known therapy.
Polymyositis
This is more common in older individuals and is associated with a progressive and symmetrical proximal muscle weakness.
On investigation, these patients have an increased creatinine kinase and are positive for anti-Jo-1 antibodies. A muscle biopsy will show muscular inflammation.
Dermatomyositis
Can be differentiated clinically from polymyositis as dermatomyositis present with cutaneous involvement. Patients with dermatomyositis may have a heliotrope rash (violaceous periorbital rash) and the “shawl sign” (where there is a rash where a shawl would hang, around the shoulders and back). Patients can also have a dorsal rash on their hands.
Remember they will also have progressive proximal muscle weakness, as in polymyositis.
On investigation, these patients have an increased creatinine kinase and are positive for anti-Jo-1 antibodies. A muscle biopsy will show muscular inflammation.
Treatment of Polymyositis/dermatomyositis
Can be responsive to steroid treatment. A high dose of corticosteroids can be given; then decrease the dose until a working maintenance dose is given. Other drugs like Azathioprine and methotrexate also have some effect.
Prognosis of Inflammatory Myopathies
Dermatomyositis responds well to therapy, one third of cases with juvenile onset recover, other third have the chances of remission, and remaining third lead life with chronic inflammation.
Inclusion body myositis (IBM) is most resistant type and is unaffected by any of the therapy.
Polymyositis has a variable prognosis. Some of the cases respond well to the therapy while some progress to chronic inflammation or may have severe disease leading to disability.
Endocrine Myopathies
Thyrotoxic myopathy develops due to the overproduction of thyroid hormone.
Hypothyroid myopathy develops due to the underproduction of thyroid hormone.
Steroid myopathy can either be endogenous or exogenous.
Clinical Features of Endocrine Myopathies
- There is muscular weakness with or without muscular pain that is more proximal than distal and it occurs in symmetric pattern
- Muscular cramps
- Muscular atrophy may or may not be present
Thyrotoxic myopathy
This can often be subclinical and patients may not know they have the condition. Patients usually have brisk reflexes (as they have thyrotoxicosis). The creatinine kinase is often normal on an investigation.
Hypothyroid myopathy
Patients can present with a proximal weakness. They may complain of fatigue or muscle pain and cramping. They have relaxation of the reflexes at an advanced stage of the disease. On investigation, these patients will have a raised creatinine kinase.
Steroid myopathy
On investigation, these patients typically have a normal creatinine kinase.
Drug/toxin-induced Myopathies
There are many toxins and drugs that cause myopathies – so many that it is impossible and impractical to list all of them. HMG-CoA reductive inhibits, or statins are one group that are commonly questioned. Others to know are:
- Fluorinated glucocorticoids (dexamethasone)
- Zidovudine (AZT) – this HIV drug can cause a mitochondrial myopathy
- Cimetidine causes inflammatory myopathy



{kind=link}
{kind=link}
{kind=link}
Comentários
Enviar um comentário